
Amino App Pc Download Archives

Amino App Pc Download Archives
AliView
About
AliView is yet another alignment viewer and editor, but this is probably one of the fastest and most intuitive to use, not so bloated and hopefully to your liking.
The general idea when designing this program has always been usability and speed, all new functions are optimized so they do not affect the general performance and capability to work swiftly with large alignments. The speed in rendering even makes it possible to work with large alignments on older hardware.
A need to easily sort, view, remove, edit and merge sequences from large transcriptome datasets initiated the design of the program.
It is of course also working very well with smaller datasets:)
The program is developed at the department of Systematic Biology, Uppsala University, so there is probably a predominance in functionality supporting those working with phylogenies.
Citation: Larsson, A. (2014). AliView: a fast and lightweight alignment viewer and editor for large data sets. Bioinformatics30(22): 3276-3278. http://dx.doi.org/10.1093/bioinformatics/btu531
The program is released under the Open Source Software License 'GNU General Public License, version 3.0 (GPLv3)'
Source code is available at GitHub:https://github.com/AliView/AliView
Download the latest stable version: 1.26 (07/Apr/2019) (Mac OS X, Windows, Linux)
Feature list:
- fast and light-weight
- simple navigation, mouse-wheel zoom out unlimited to see whole alignment, and zoom in on place of interest
- various visual cues to highlight consensus characters or characters deviating from the consensus or characters deviating from a selected "trace"-sequence
- edit sequences/alignment (manually), insert, delete, change, move, rename (with keyboard or mouse)
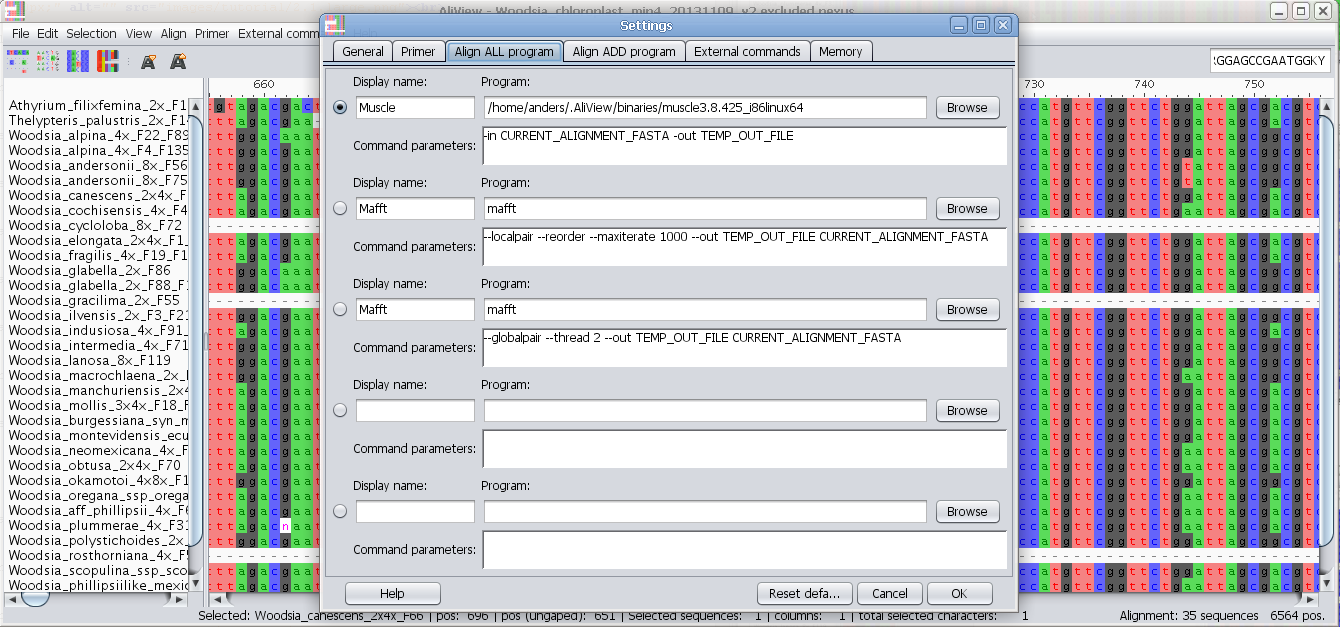
- align, add and align automatically with MUSCLE (included) or MAFFT, or any other aligner of your choice
- define aligner program presets (different parameters, different software)
- align new sequences to existing or realign all
- realign a selected block
- realign nucleotides as translated amino-acids
- delete vertical gaps
- undo/redo
- find degenerate primers in conserved regions in an alignment of mixed species
- open and save in FASTA-, NEXUS-, PHYLIP, CLUSTAL or MSF-format (unlimited file sizes)
- print (current view)
- export whole alignment as an image file (png-format)
- copy selection (as fasta-sequences or just characters)
- paste sequences (both as sequences in clipboard or filenames in clipboard)
- add more sequences from file
- merge two sequences - and calculate consensus if the overlap is not perfect
- translate (view) nucleotide sequences as amino-acid sequences
- save translated alignment
- read and preserve Codonpos, Charset and Excludes (Nexus-specification)
- change Codonpos for selected regions (Nexus-specification)
- drag-drop/remove of sequences/files
- move sequences to top/bottom with key-stroke
- a very simple to use "external interface" that lets you invoke your other favorite programs (you could for example automatically have the alignment sent to FastTree and then automatically opened in FigTree)
- search function that finds patterns across gaps and follows IUPAC codes
- search multiple sequence names at once (stored in clipboard)
- reverse complement/reverse/complement sequences or whole alignment
- different color schemes including ClustalX
- sort sequences by name
- sort sequences by residue in selected column
Illustrated feature-list:
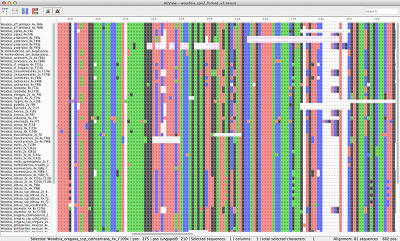
Unlimited mouse wheel zoom in/out to see whole alignment
Speedy handling of large alignments
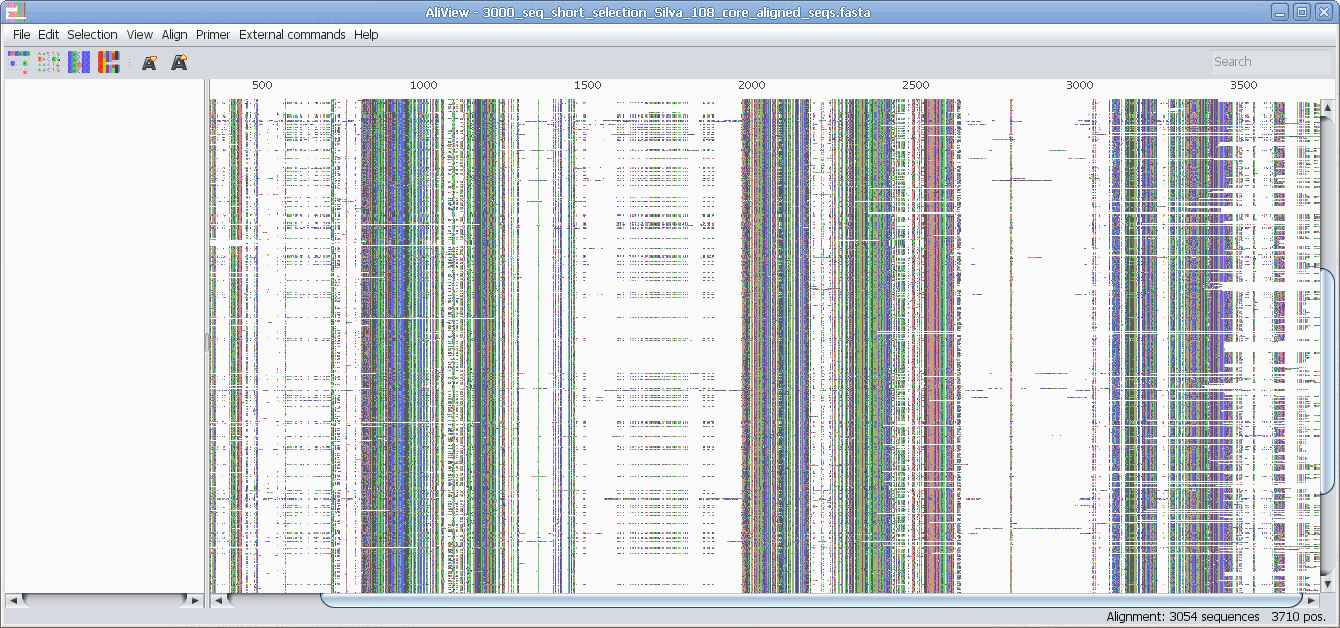
Reads unlimited size FASTA, PHYLIP, NEXUS, CLUSTAL and MSF files
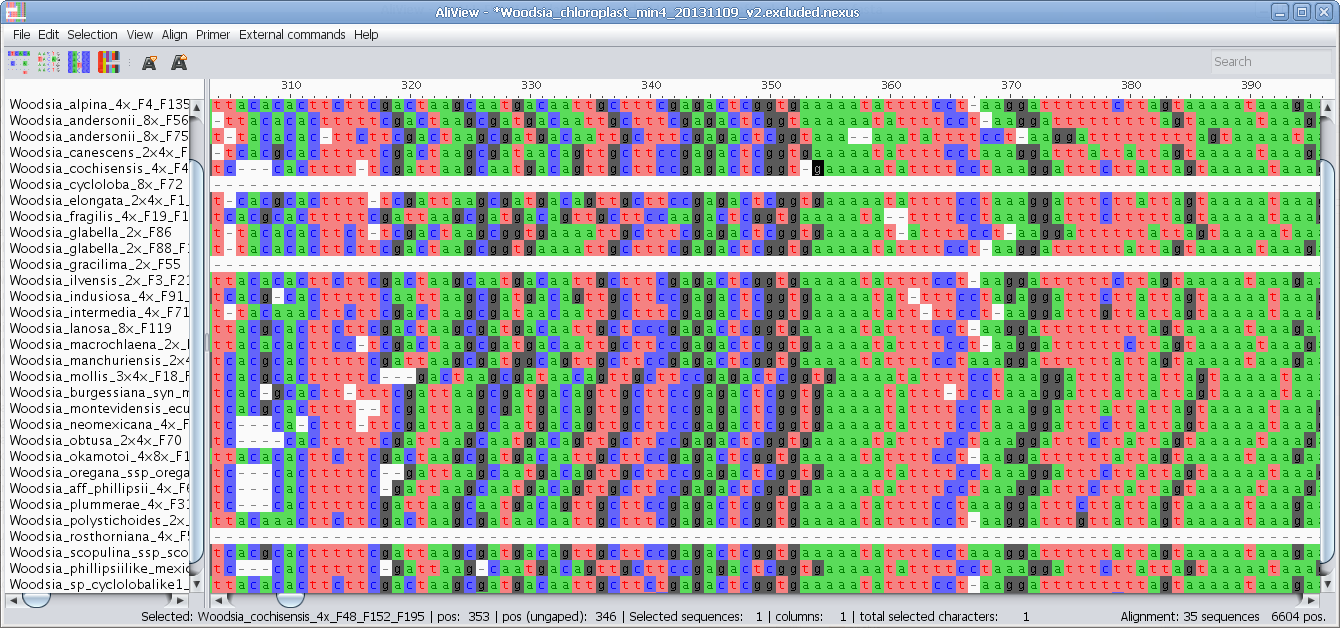
Alternative Color schemes (SeaView)
ClustalX consensus color scheme
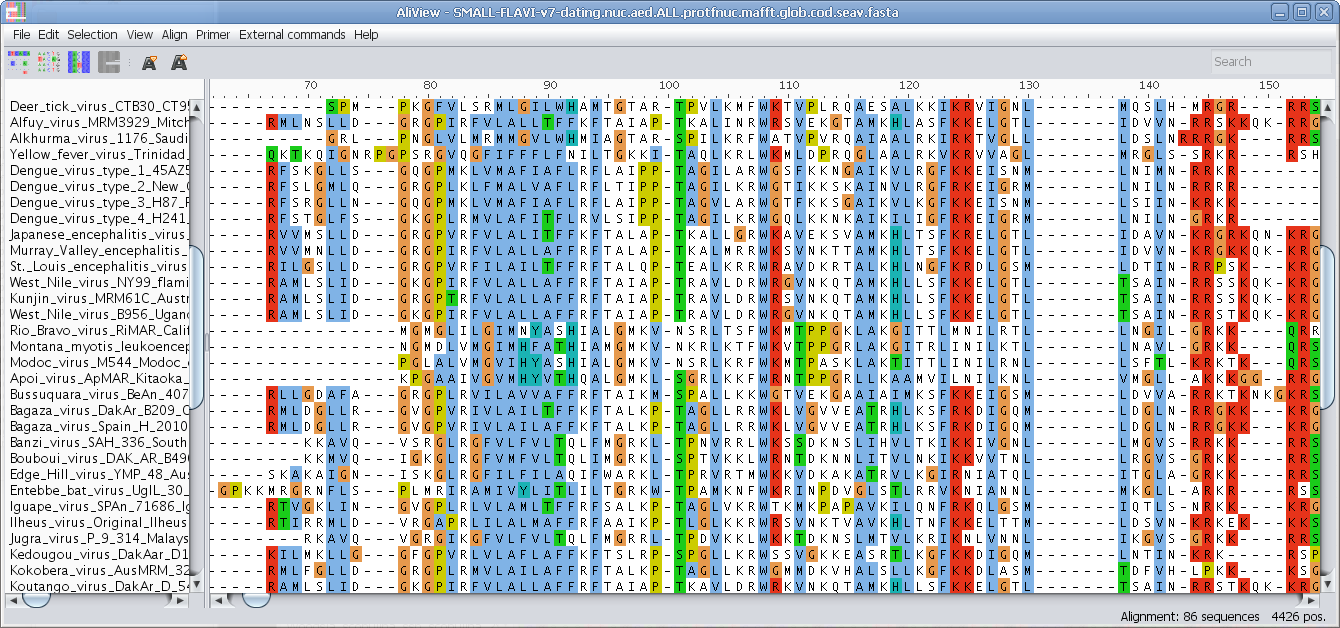
View nucleotides colored as translated Amino Acids
View nucleotides colored as translated Amino Acids (1-position)
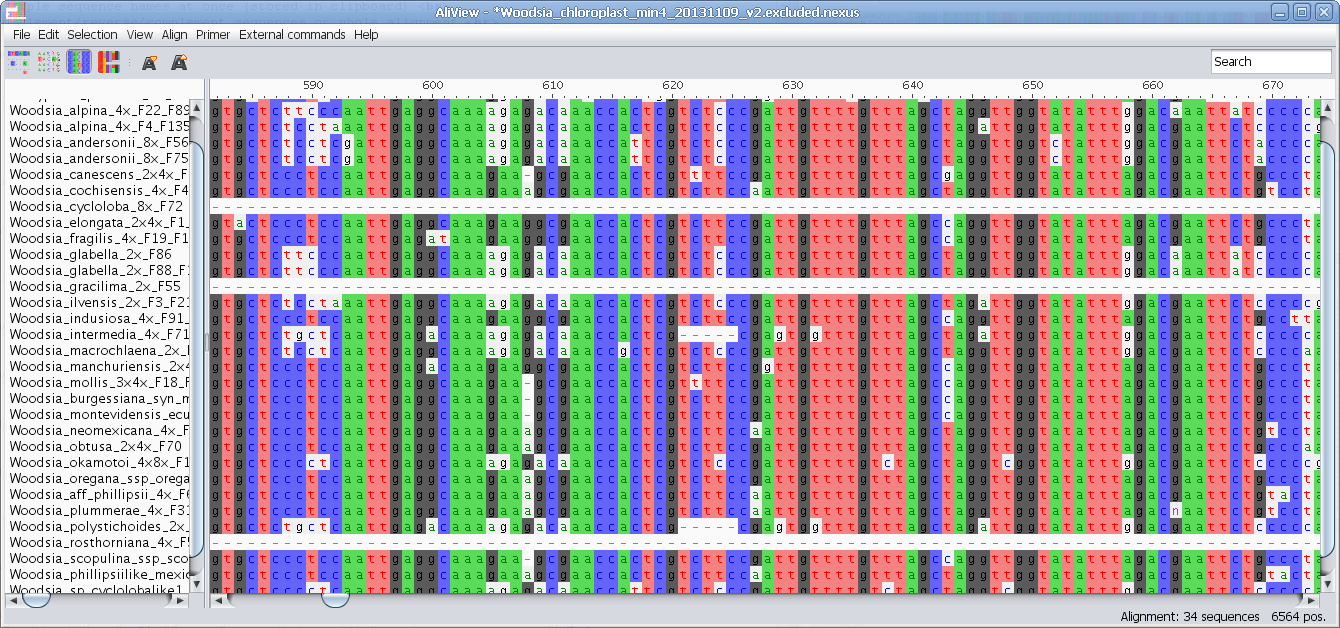
Highlight characters deviating from Consensus
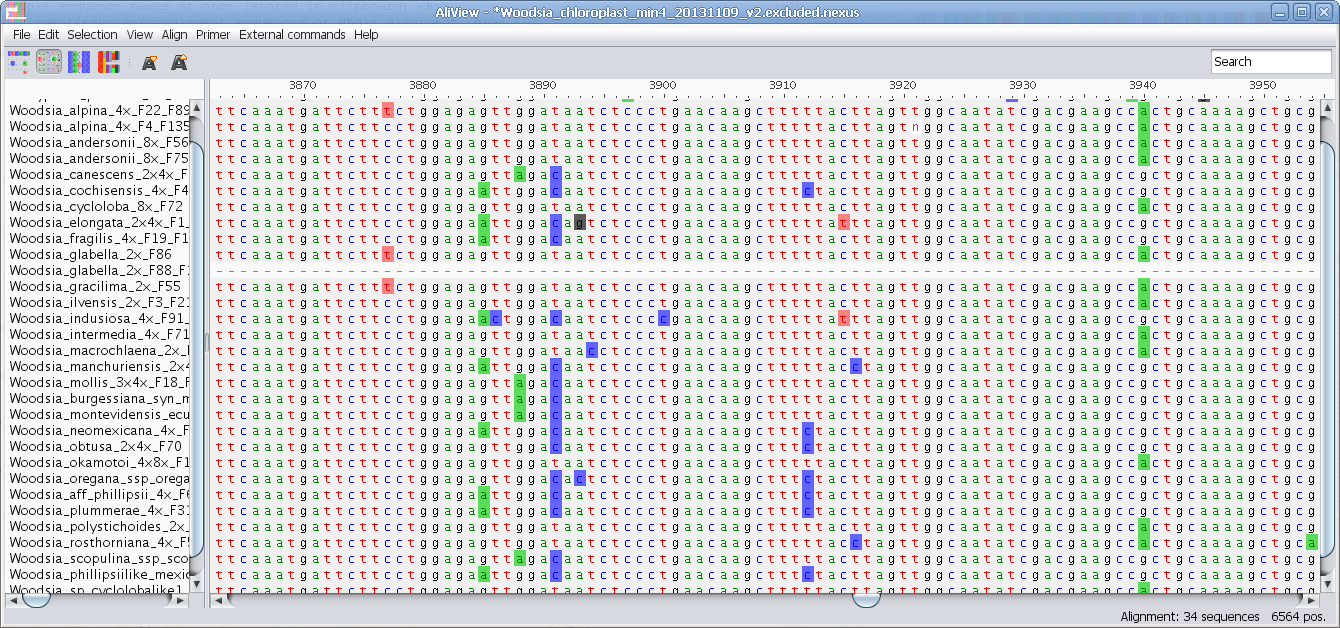
Highlight characters deviating from selected "Trace" sequence
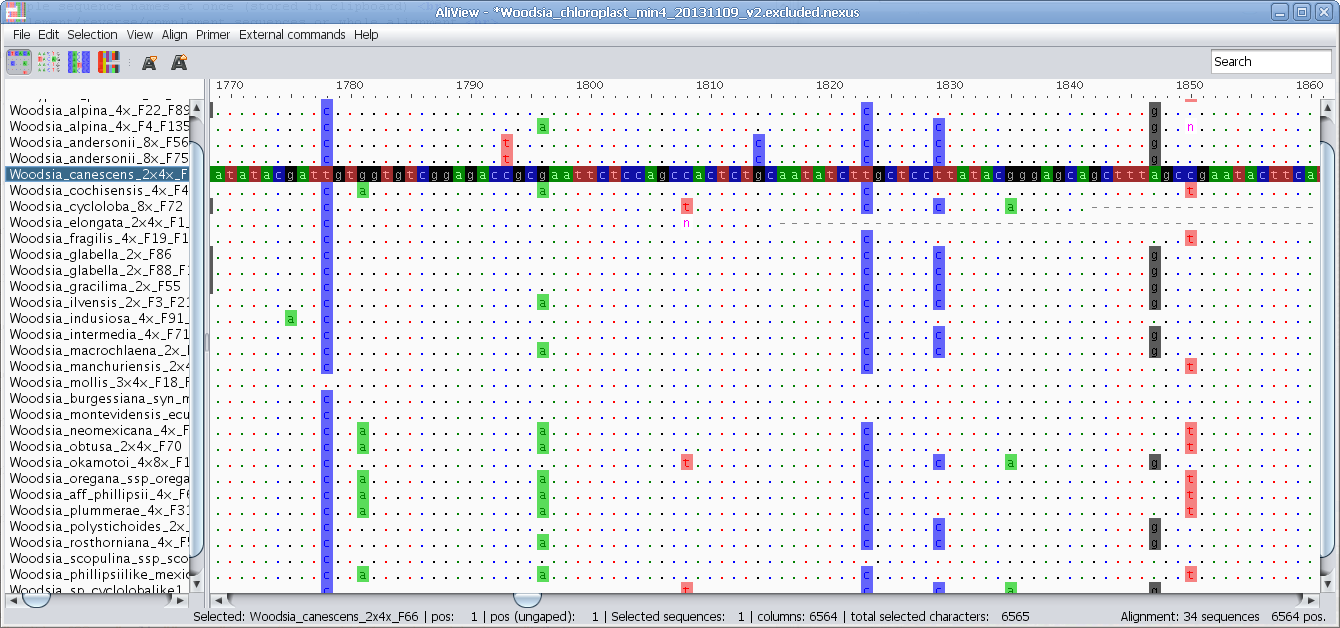
Highlight characters belonging to Consensus
MUSCLE or other alignment program to realign sequences
Realign selected block with muscle or other aligner program
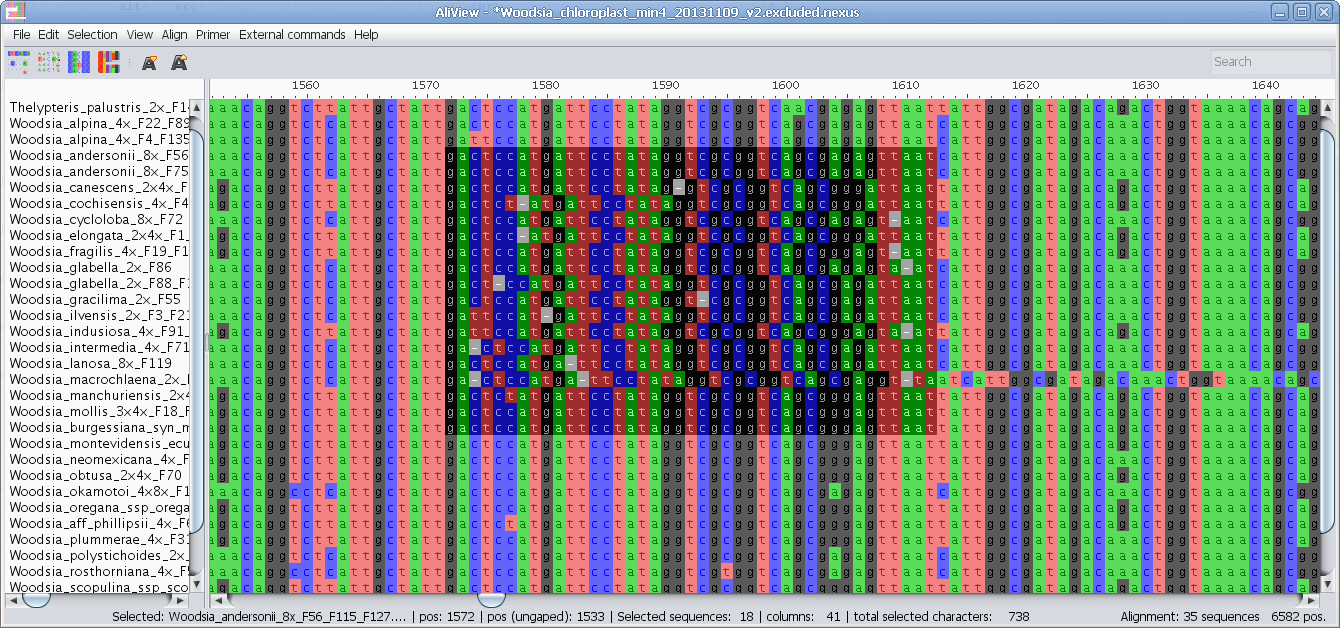
Realign single sequence with MUSCLE or other aligner program
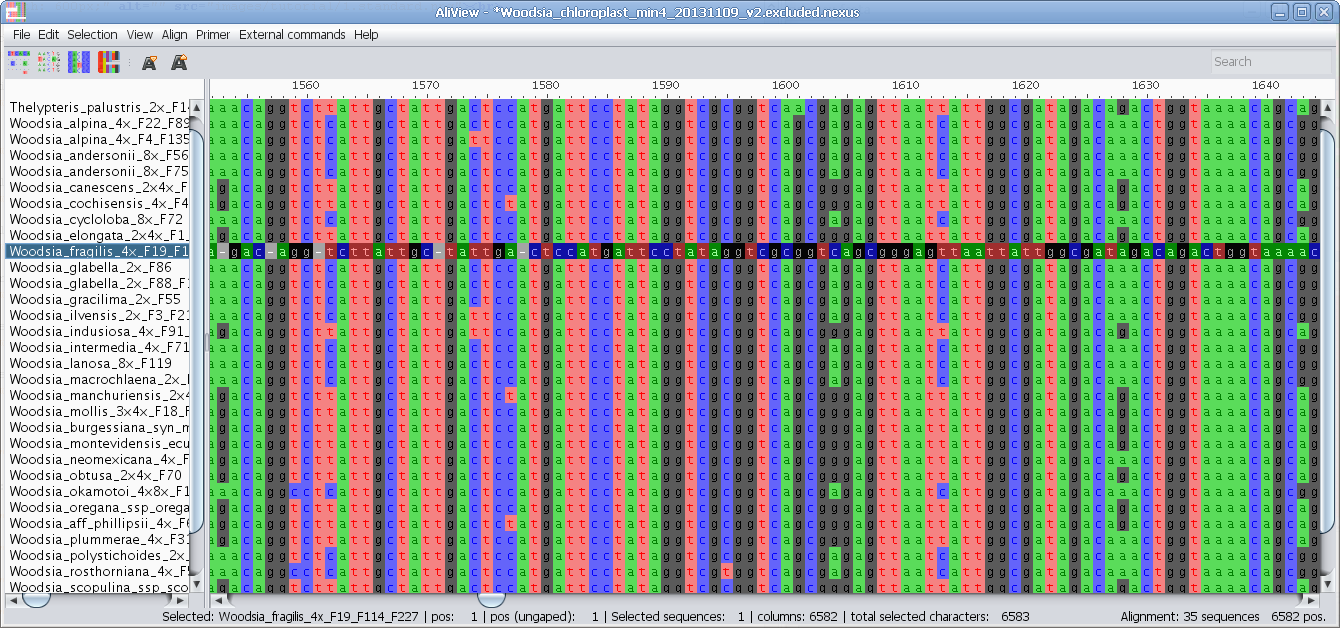
Nucleotides realigned as translated AminoAcids and then retranslated to nucleotides
Manual Alignment with Keyboard or Mouse
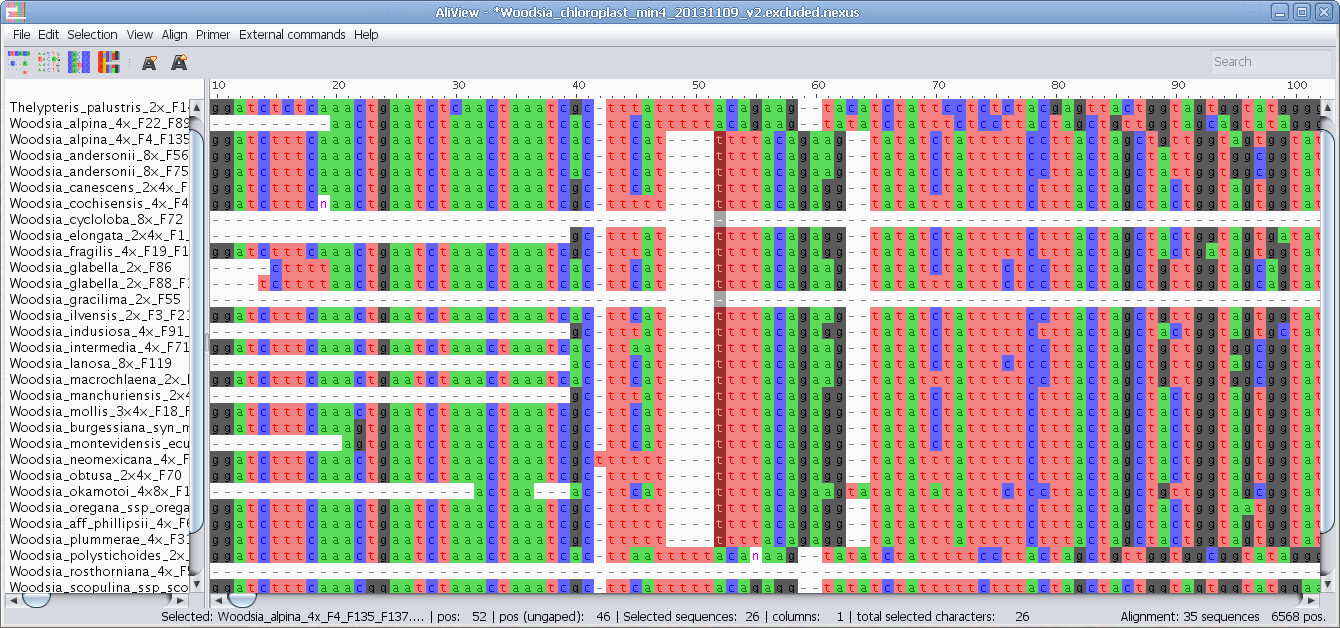
Delete sequences, reorder, move to top, move to bottom
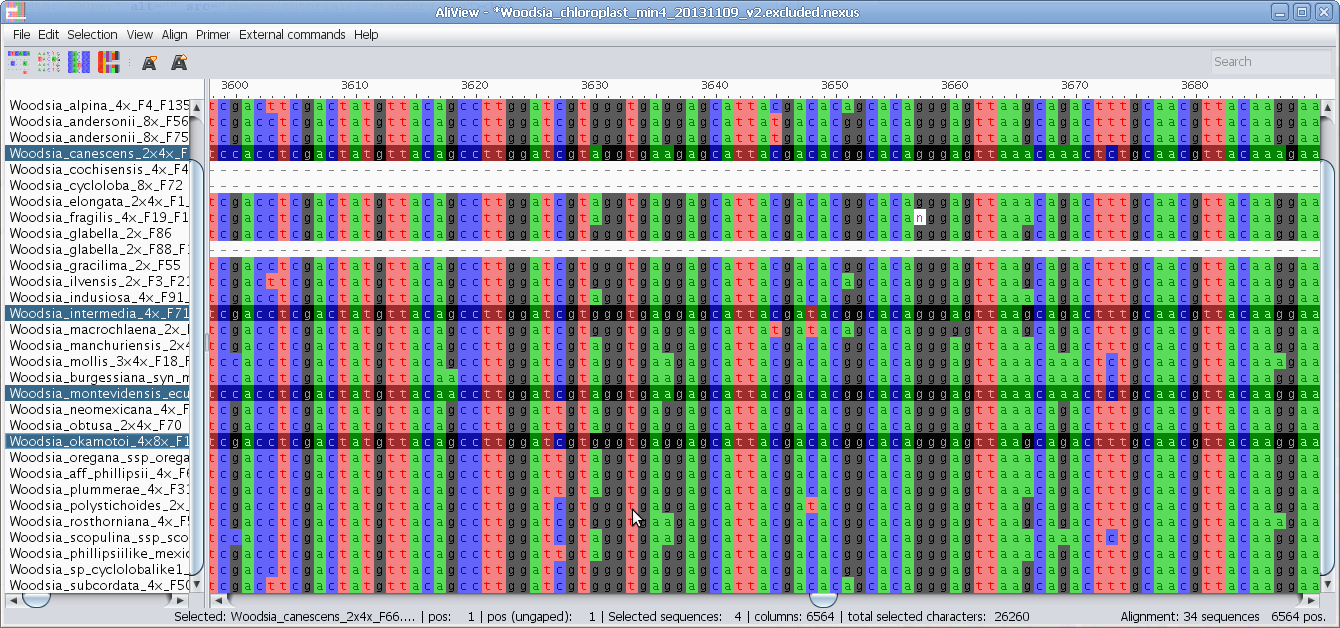
Merge sequences (overlapping or not - create consensus if overlapping)
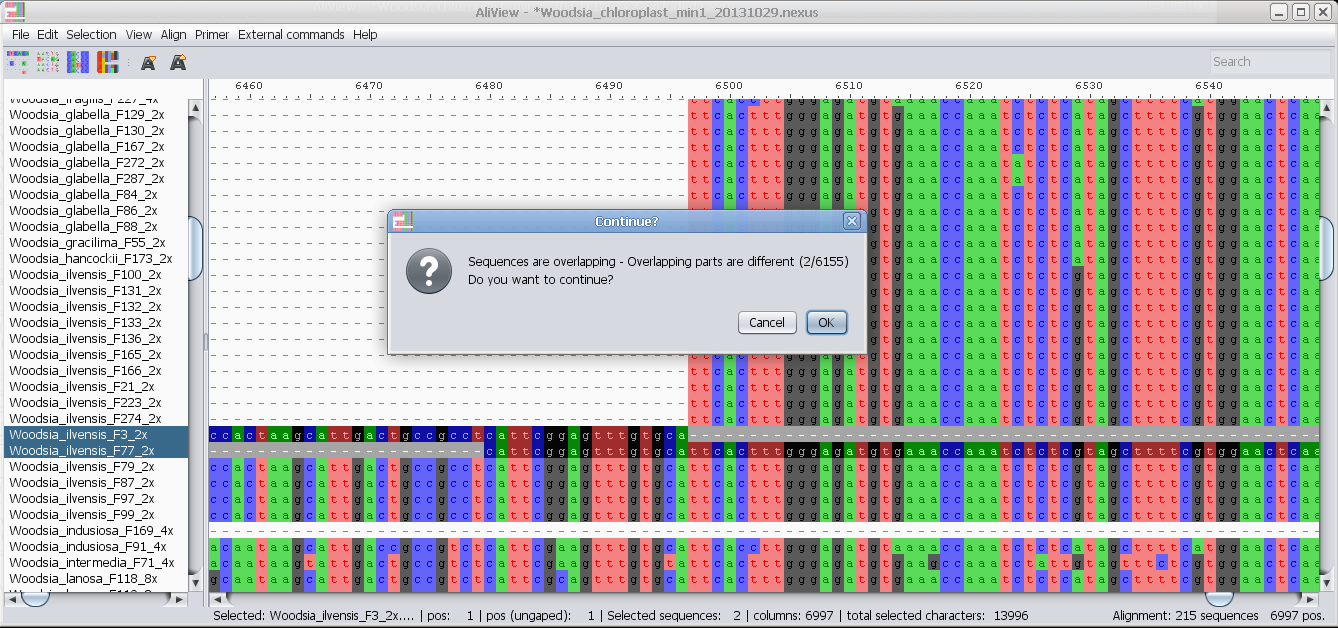
Drag and drop file/sequences, if you hold Shift when dropping
then you add to current alignment, otherwise open new
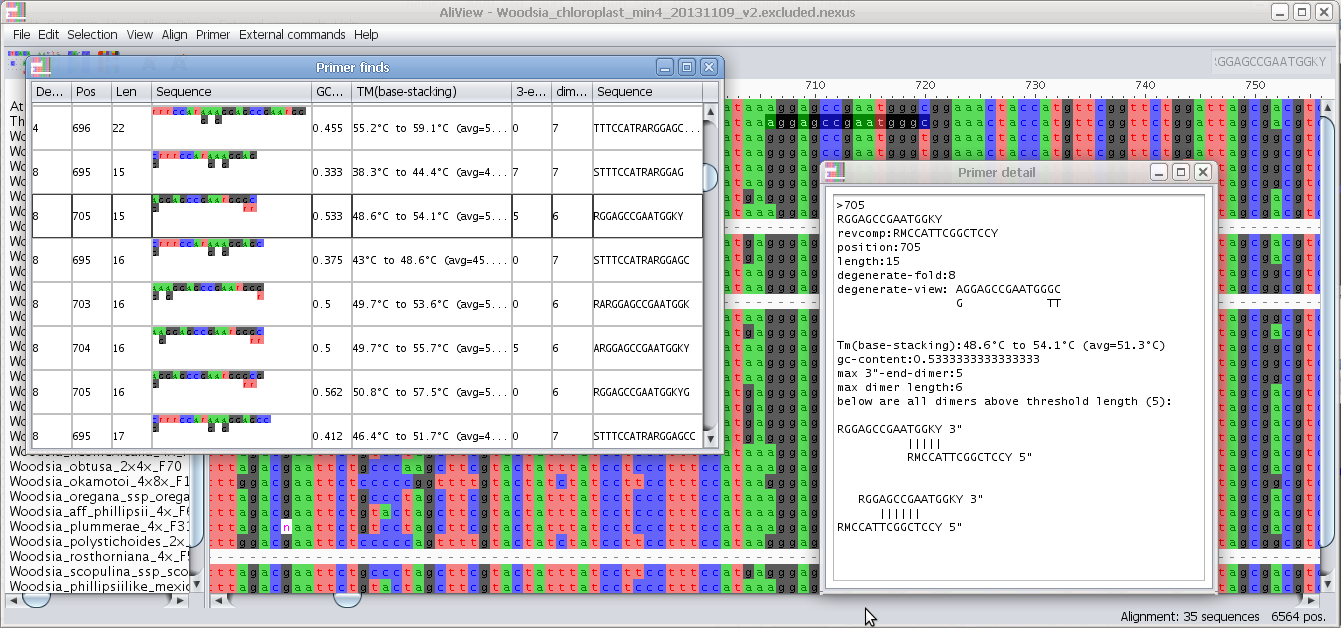
Find degenerate primers in selected blocks
Configure external programs
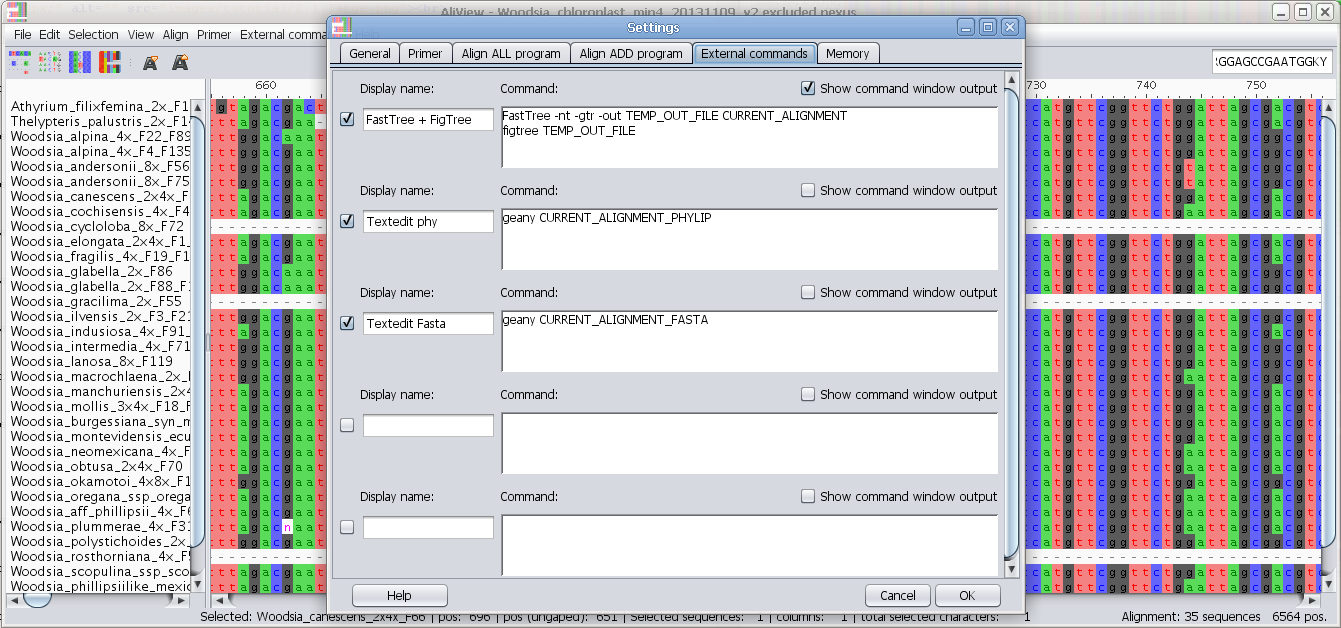
Add and configure other alignment programs to use e.g. MAFFT
Install instructions and Downloads:
Version history here (version_history.txt)
Some demo datasets can be found here
Mac OS X (all versions) download here
The program is distributed as an Application (AliView-app.zip), that only needs to be dropped in your App-folder (sometimes you need to unpack it manually)
OBS! On Mavericks or Sierra you might get an error message ”This application is damaged and can't be opened. You should move it to the Trash.” - The file is actually not damaged, it is just a bad error message. There is a settings in the ”Gatekeeper” that you need to change to be able to download software from other places than the App-store.
How to solve (on Mavericks): http://answers.uchicago.edu/page.php?id=25481
How to solve (on Sierra): https://www.tekrevue.com/tip/gatekeeper-macos-sierra
Windows (all versions) download here
Download and run AliView-Setup.exe
- the setup program will default installation to directory
Alternatively you can download the program AliView.exe or aliview.jar from sub-directory
NOTE: some virus scanners might block the installation and need to be temporarily disabled.
Linux (all versions) download here
The simplest install is to download the file: aliview.install.run
(this is a executable archive that will copy the files to the /usr/bin/ and /usr/share/aliview/, and will also install a ".desktop" link to the start-menu on compatible systems.)
- after downloading you will likely need to change the execution rights of the install file:
- install by issuing command:
(most likely you need to sudo the installation, if you don't have super user rights to the system see below)
- start the program with command:
(this is a sh-script that will execute java -jar aliview.jar)
Linux (alternative installation - as non super user)
1. Download the archive aliview.tgz and extract it into a directory of your choice.
2. Run program from this directory by issuing command in the terminal
(this is a sh-script that will execute command java -jar aliview.jar)
NOTE: If you are running Linux, consider upgrading to Java 8, there is a significant improvement in drawing speed since it is using Linux built in XRender.
Installation is very simple (1 minute) - Java 8 installation instructions for Ubuntu/Debian/OpenSuse/RedHat
Report a bug or feature request:
Two ways:
1. Report issue at AliView GitHub Issues tracker: https://github.com/AliView/AliView/issues/
or
2. Send email to: anders.larsson [at] icm.uu.se
Contact:
Anders Larsson,
NBIS, Department of Cell and Molecular Biology
Uppsala University
Sweden
email: anders.larsson [at] icm.uu.se
Citation:
Larsson, A. (2014). AliView: a fast and lightweight alignment viewer and editor for large data sets. Bioinformatics30(22): 3276-3278. http://dx.doi.org/10.1093/bioinformatics/btu531
Contents:
File menu
Edit menu
Selection menu
View menu
Align menu
Tools menu
Large files indexed
Memory settings
General setting (Preferences window)
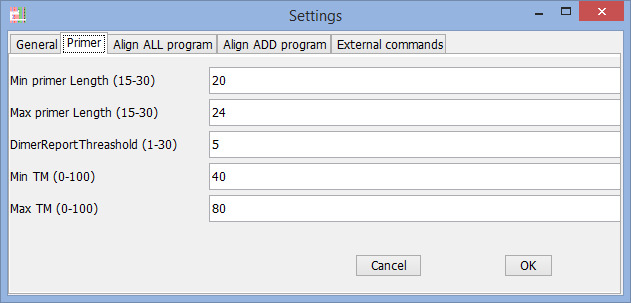
Find primer setting (Preferences window)
Align All Commands - Edit alignment program
Align Add Commands - Edit alignment program
External Commands - Add and edit programs called by AliView
Drag and drop tips
File menu:
Open File: AliView will open files in Fasta, Fastq, Nexus, Phylip, Clustal and MSF format.
Save as Nexus simplified names: Unorthodox characters (- \?./|\,&) in Sequence names are replaced by "_"
Save as Codonpos Nexus (nucleotide dataset only): All pos-1 nucleotides are saved as one charset in the beginning of the alignment, thereafter all pos-2 nucleotides as one charset, then all pos-3 and finally all non-coding (0-pos).
Print: Apart from printing to paper, it is often possible in the settings to "print to file", this way you can get post script (ps) files of the alignment.
Export alignment as image: This will save the entire alignment as a .png image file
Save selection as Fasta: Only the selected sequences, or selected parts of sequences will be included in the .fasta file - a very simple way of pruning away things from your alignment.
Save Translated alignment as Amino acids (nucleotide dataset only): Program will translate nucleotides into AminoAcids (taking into account user specified edits in codon positions when translating)
Start debug: Prints more messages
Show message log: Displays program messages and errors in a message window
Edit:
When opening very large files and they are indexed, then the editing capabilities are very limited - but if you need to edit it might be possible if you increase the Memory resources for AliViev see here.
Undo/Redo: AliView has undo/redo functionality (except when large indexed files are read from file). Depending on the Memory settings, the number of possible undo/redo steps changes.
Copy selection as Fasta: The selected sequences/characters are copied into clipboard as a Fasta sequences (including both name and sequence).
Copy selection as characters: The selected sequences/characters are copied into clipboard as characters only - Not including the names of the sequences where the characters come from.
Paste (fasta sequences): Sequence(s) will be pasted at top of alignment if clipboard contains either File containing fasta sequences or text in fasta format, e.g.:
>Name_of_seq
TTGGGCATG
Add sequences from file: Add sequence(s) from a file to be pasted at top of alignment (FASTA,PHYLIP, NEXUS, CLUSTAL or MSF file format).
Edit mode: It is not possible to edit alignment by accident unless you activate "Edit Mode".
Clear selected bases: Changes selected nucleotides/AminoAcids into GAP.
Delete selected Removes selected sequences/nucleotides/AminoAcids from alignment.
Delete selected bases: Removes selected nucleotides/AminoAcids from sequences/alignment.
Delete vertical gaps: If there are column(s) in the alignment only containing GAPs these positions are removed.
Delete all gaps in all sequences: Degap the whole alignment.
Trim sequences/alignment: If there are column(s) in the beginning or at the end of the alignment containing GAPs only, then these positions are removed.
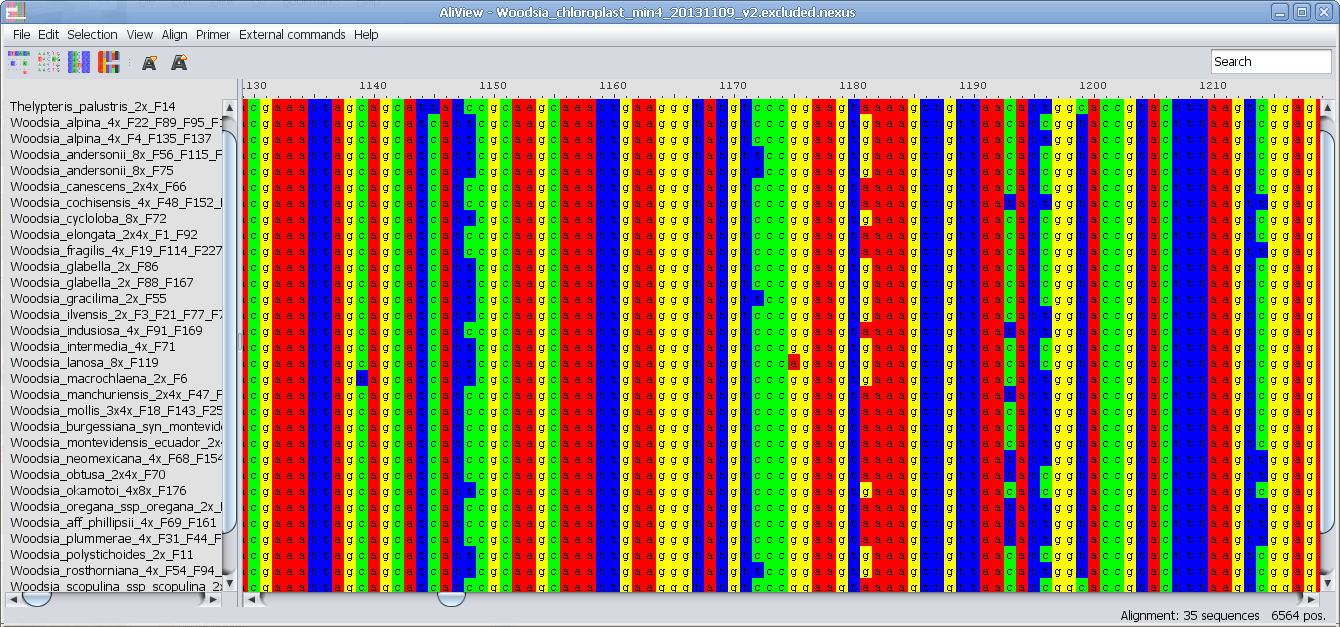
Excludes (Nexus specification) can be useful if you have an alignment with sections that you don't want to remove permanently - but sometimes when you make an analysis. Then you can save the alignment with the "Excluded" sections removed and use that alignment for the analysis.
Delete excluded bases: Removes sections of the alignment that are marked as "Excluded" (Nexus specification). See also above.
Replace terminal GAPs into missing char (?)
Replace missing char (?) into GAP (-)
Find Finds in name or nucleotides/aminoacids - searches across gaps and obeys IUPAC codes.
Find sequence names from clipboard Finds and selects sequence(s) matching names that are stored in the clipboard as a list.
Example list in clipboard:
Woodsia
Athyrium
Dryopteris
Merge two selected sequences: If there is a non-identical overlap AliView will create a consensus sequence.
Reverse complement clipboard: Will reverse complement characters/fasta sequences that are stored in clipboard.
Selection:
Select all
De-select
Expand selection Right
Expand selection Left
Expand selection Down
Expand selection Up
Move selected sequences up
Move selected sequences down
Move selected sequences to top
Move selected sequences to bottom
Add selection to Excludes: Marks sections of the alignment as "Excludes" (Nexus specification). See also above under Excludes (this is saved in the NEXUS Excludes block).
Remove selection from Excludes: Removes selected sections of the alignment from "Excludes" (if they are included already). See also above under Excludes (Nexus specification).
Select charset: If the alignment file has charsets defined you can select one.
Set selection as coding: In a nucleotide alignment you can specify the coding regions, pick the start position of the selection and then further bases will be marked as codon position 1,2,3,1,2,3... and so forth. (this is saved in the NEXUS Codonpos block)
Set selection as Non-coding: Set bases as non-coding (this is saved in the NEXUS Codonpos block)
View:
Decrease Font Size
Decrease Font Size
Highlight consensus characters Characters in a column that belong to majority rule consensus are highlighted by removing background colors on deviating characters.
Tools:
Primer:
Find primer in current selection:
Note - You can change the essential parameters when searching for primers in the Find primer settings
1. Select a block in the Alignment where you want to find a primer.
2. Select the menu "Find primer in current selection"
3. AliView will now open a window with all possible primers in the selected region (min length and max length of the primer is specified in "Find primer settings") as an ordered list sorted by the number of degenerate positions, self-binding values and melting temperature (TM).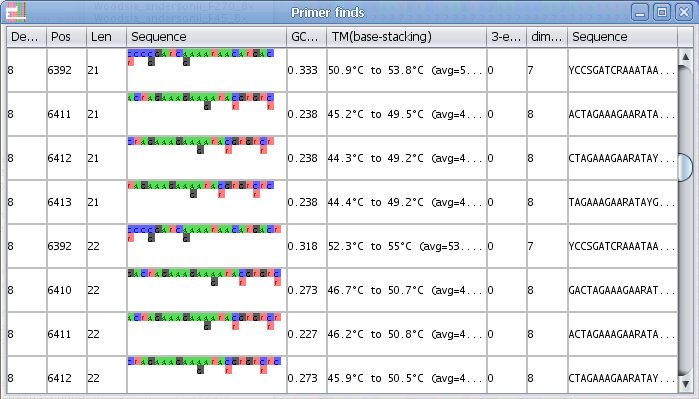
4. If you click on a specific primer another window will open with all primer details - and the primer position will also be highlighted in the alignment.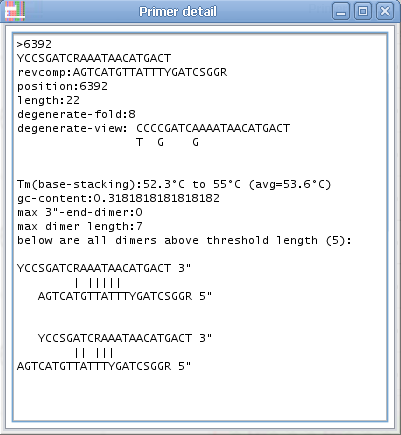
External commands:
Edit external commands: You can have AliView call other programs with the current alignment as a parameter see here for examples how to design a command
Help menu
Help AliView will open this document from this menu.....
Load more sequences from file
If you have opened a large file and all sequences were not indexed at once, this menu will show up and you can select other parts of the file to index. You can also change the number of sequences to index at once in the Program preferences, General settings
Memory settings
If you want AliView to read larger alignments in memory and not from file (this allows for more editing capabilities), then you can change the maximum memory settings for the program.
The amount of memory needed for a file to be read into memory is about 2 x file size.
Mac OS X
Go to Applications in Finder -->Left click AliView --> Show Package Content --> Contents --> Then open the file "Info.Plist" in a text-editor and change the parameter: <string>-Xmx512m -Xms128m</string> to something different (for example 2GB=2048M):
<string>-Xmx2048m -Xms128m</string>
Linux
/usr/bin/aliview
open this file in text-editor and change the parameter -Xmx1024M (default setting = 1024M memory)
Windows
In the installation folder of AliView (default: "c:\Program Files\AliView\") open the file "AliView.l4j.ini" in a text editor and change the setting: -Xmx1024m to something you prefer (for example 2GB=2048M) -Xmx2048m
General settings
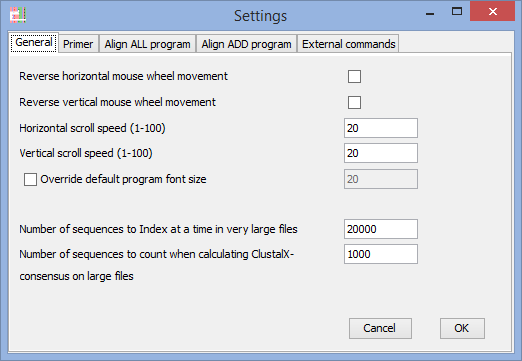
Reverse horizontal mouse-wheel (touchpad) movement
Reverse vertical mouse-wheel (touchpad) movement
(if the direction if movement is driving you crazy - adjust it here).
Horizontal scroll speed
Vertical scroll speed
(on some systems the movement is to fast/slow) – you can adjust it here.
Override default program font size
(on some computers with extreme resolution it might be hard to read text with default settings - OBS! If you are having these problems on Windows, then you should consider installing Java 9, dpi-scaling works on Windows with Java 9)
When working with large files being read straight from disk:
Number of sequences to index at a time in very large files
(you can change the number of sequences being indexed at once – the rest of the sequences in the file are available by selecting the menu-alternative ”Load more sequences” (this menu is only showing up when the file is parted into ”pages”). More sequences are being indexed as they are being viewed.
Number of sequences to count when calculating ClustalX consensus on large files
(it would take very long time to calculate consensus considering every position in a large file)
Find Primer settings
Program to use for "Align all" commands
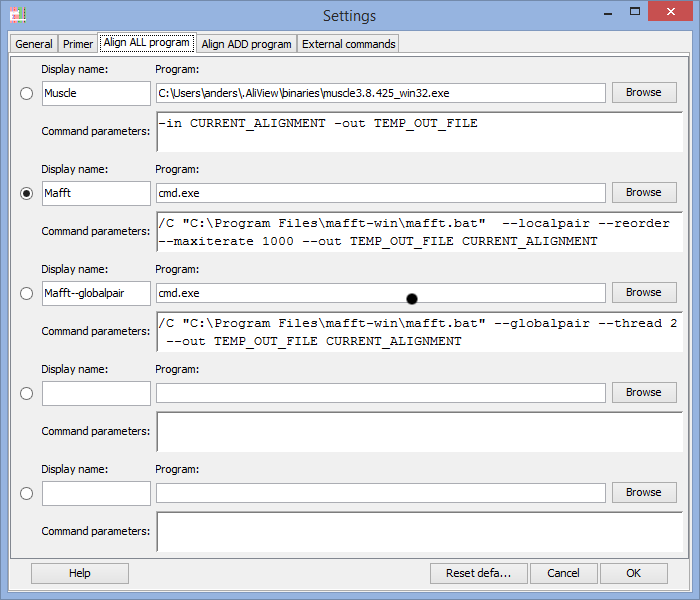
In the Program field you enter the program you want to execute when using the alignment functions in AliView.
There are default settings for MUSCLE and MAFFT included in AliView and they look different for different operating systems (Windows / Linux / Mac OSX).
Below are commented examples for Mac OS X, Linux and Windows
Note 1:It is essential that file names that contains spaces or unorthodox characters are specified within quotation marks "
On Windows for example: "c:\Program Files\mafft-win\mafft.bat"
On Mac OS X for example: "FigTree 1.4.0.App"
Note 2: When calling MAFFT on Windows computers you are actually starting the program cmd.exe and instead including MAFFT program path as a parameter (compare for example with the muscle.exe program and command).
Program to use when adding and aligning in new sequences ("Align add" commands)
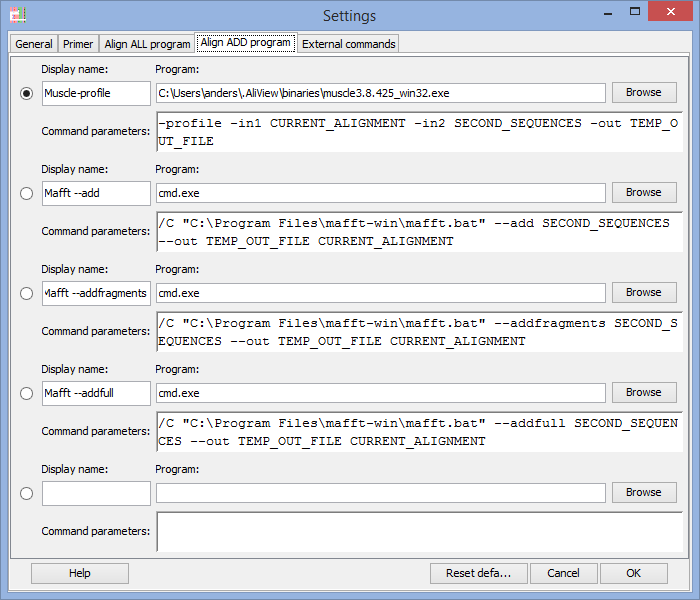
For an explanation on how to fill in the fields see above "Program to use for "Align all" commands" and below are also commented examples for Mac OS X, Linux and Windows
Some commented examples of Aligner Program commands:
NOTE: It is essential that file names that contains spaces are specified within quotation marks "MAC OS X
| Align ALL command examples | |
| MUSCLE command (default) | Comment |
| The alignment program (MUSCLE) is run with parameter -in -out AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will reload the output file from alignment program TEMP_OUT_FILE | |
| MAFFT example | Comment |
| NOTE that MAFFT is not installed with AliView, and has to be installed separately In this example the program (MAFFT) is run with parameter --localpair --reorder --maxiterate 1000 --out AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will automatic reload the output file from alignment program TEMP_OUT_FILE | |
| Align ADD command examples | |
| MUSCLE command (default) | Comment |
| The alignment program (MUSCLE) is run with parameter -profile -in1 -in2 -out This is the MUSCLE way of adding sequences to an existing alignment. AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text SECOND_SEQUENCES with a temporary file in which AliView saves the new sequences. AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will reload the output file from alignment program TEMP_OUT_FILE | |
| MAFFT example | Comment |
| NOTE that MAFFT is not installed with AliView, and has to be installed separately In this example the program (MAFFT) is run with parameter --add --out AliView will replace the text SECOND_SEQUENCES with a temporary file in which AliView saves the new sequences. AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will automatic reload the output file from alignment program TEMP_OUT_FILE | |
WINDOWS
| Align ALL command examples | |
| MUSCLE command (default) | Comment |
| The alignment program (MUSCLE) is run with parameter -in -out AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will reload the output file from alignment program TEMP_OUT_FILE | |
| MAFFT example | Comment |
| NOTE that MAFFT is not installed with AliView, and has to be installed separately In this example the program (MAFFT) is run with parameter --localpair --reorder --maxiterate 1000 --out NOTE2 The program mafft.bat is actually called by program cmd.exe with parameter /C AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will automatic reload the output file from alignment program TEMP_OUT_FILE | |
| Align ADD command examples | |
| MUSCLE command (default) | Comment |
| The alignment program (MUSCLE) is run with parameter -profile -in1 -in2 -out This is the MUSCLE way of adding sequences to an existing alignment. AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text SECOND_SEQUENCES with a temporary file in which AliView saves the new sequences. AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will reload the output file from alignment program TEMP_OUT_FILE | |
| MAFFT example | Comment |
| cmd.exe | NOTE that MAFFT is not installed with AliView, and has to be installed separately NOTE2 The program mafft.bat is actually called by program cmd.exe with parameter /C In this example the program (MAFFT) is run with parameter --add --out AliView will replace the text SECOND_SEQUENCES with a temporary file in which AliView saves the new sequences. AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will automatic reload the output file from alignment program TEMP_OUT_FILE |
LINUX
| Align ALL command examples | |
| MUSCLE command (default) | Comment |
| The alignment program (MUSCLE) is run with parameter -in -out AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will reload the output file from alignment program TEMP_OUT_FILE | |
| MAFFT example | Comment |
| NOTE that MAFFT is not installed with AliView, and has to be installed separately In this example the program (MAFFT) is run with parameter --localpair --reorder --maxiterate 1000 --out AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will automatic reload the output file from alignment program TEMP_OUT_FILE | |
| Align ADD command examples | |
| MUSCLE command (default) | Comment |
| The alignment program (MUSCLE) is run with parameter -profile -in1 -in2 -out This is the MUSCLE way of adding sequences to an existing alignment. AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text SECOND_SEQUENCES with a temporary file in which AliView saves the new sequences. AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will reload the output file from alignment program TEMP_OUT_FILE | |
| MAFFT example | Comment |
| NOTE that MAFFT is not installed with AliView, and has to be installed separately In this example the program (MAFFT) is run with parameter --add --out AliView will replace the text SECOND_SEQUENCES with a temporary file in which AliView saves the new sequences. AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer When the alignment program is done - AliView will automatic reload the output file from alignment program TEMP_OUT_FILE | |
External commands - Send the alignment to other programs
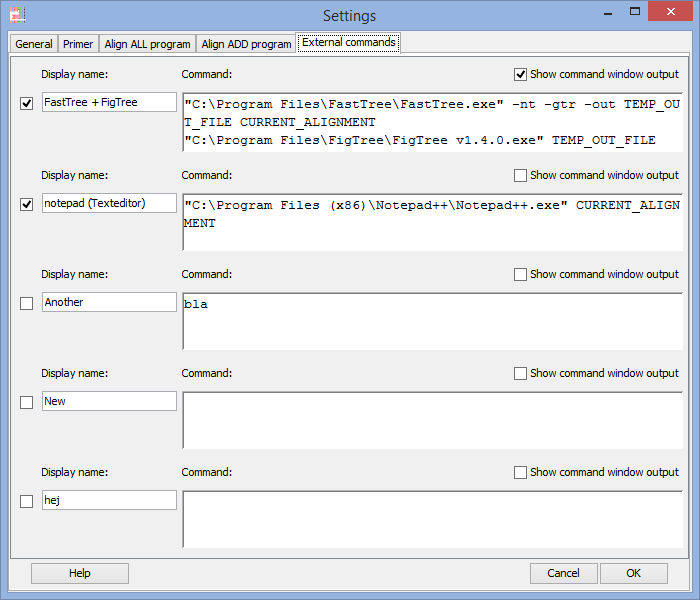
You can send the active alignment from AliView as a command parameter to other programs and create a simple pipeline of commands executed from a Menu-button in AliView.
Each line is executed as a separate command waiting for the previous to have finished. An an example of this would be a simple combination of commands that take the current alignment and run it in FastTree to create a Maximum Likelihood Tree and finally open the resulting tree in FigTree.Special Parameters:
CURRENT_ALIGNMENT_FASTA = The current alignment as a fasta file
CURRENT_ALIGNMENT_PHYLIP = The current alignment as a phylip file
TEMP_OUT_FILE = An empty temporary file created by AliView in the temp-directory of the computer
ALIVIEW_OPEN file-name = Open a new window with file name as parameter
Some examples of external commands with comments:
NOTE: It is essential that file names that contains spaces or unorthodox characters are specified within quotation marks "| MAC OS X | |
| Command | Comment |
| NOTE that FigTree application name is surrounded with quotation marks since the name contains a space. NOTE that FigTree application is started with MacOS X command open -a The program FastTree is run with parameters -nt -gtr -out [tree-file-out] [input_alignment] AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment When FastTree.exe is finished creating the tree, it will have saved the result into the file TEMP_OUT_FILE FigTree v1.4.0 is run when FastTree is finished and as parameter to the program is the tree in the file TEMP_OUT_FILE | |
| WINDOWS | |
| Command | Comment |
| NOTE that FastTree.exe and FigTree.exe application names are surrounded with quotation marks " since the file path and name contains a space. The program FastTree.exe is run with parameters -nt -gtr -out [tree-file-out] [input_alignment] AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment When FastTree.exe is finished creating the tree, it will have saved the result into the file TEMP_OUT_FILE FigTree v1.4.0.exe is run when FastTree.exe is finished and as parameter to the program is the tree in the file TEMP_OUT_FILE | |
| LINUX | |
| Command | Comment |
| NOTE that the file name should be enclosed in quotation marks if there is space in the path. The program FastTree is run with parameters -nt -gtr -out [tree-file-out] [input_alignment] AliView will replace the text TEMP_OUT_FILE with a temporary file name created in the temp-directory on the computer AliView will replace the text CURRENT_ALIGNMENT_FASTA with a temporary file in which AliView saves the current alignment When FastTree.exe is finished creating the tree, it will have saved the result into the file TEMP_OUT_FILE FigTree v1.4.0 is run when FastTree is finished and as parameter to the program is the tree in the file TEMP_OUT_FILE | |
Drag and Drop
If you drop a file in the program window it will open a new window with the alignment
Note: If you drop a sequence while pressing the "Shift"-button - the sequences in the file will be added to the currently loaded alignment.
Java 8 JRE Install (Linux)
This is tested on Ubuntu/Debian/OpenSuse (RedHat - just download and install rpm-package)(These installation instructions are compiled and simplified fom this excellent tutorial):
http://install-things.com/2014/03/24/how-to-install-oracle-java-8-on-ubuntu-12-04-linux/
1. Download jre tar.gz-version:
http://www.oracle.com/technetwork/java/javase/downloads/jre8-downloads-2133155.html
2. Unpack
sudo tar zxvf jre-8*.tar.gz
3. Move whole unpacked dir:
4. Tell your system where new java is located:
5. Tell your system to use new version:
Java 9 JRE Install (Linux)
This is tested on Ubuntu/Debian/OpenSuse (RedHat - just download and install rpm-package) # 1. Download jre tar.gz-version:
http://www.oracle.com/technetwork/java/javase/downloads/jre9-downloads-3848532.html
# 2. Unpack
# 3. Make sure jvm-dir exists:
# 4. Move whole unpacked dir:
# 5. Tell your system where new java is located:
# 6. Tell your system to use new version:
Setup and Dependencies
This file describes the installation process for Amino.
The following dependies are required or optional. Please see below for the corresponding list of Debian and Ubuntu packages. Also, please consult the errata notes below as some package versions have missing features or bugs that require workarounds.
Required Dependencies
These dependecies are required to compile and use Amino:
Optional Dependencies
These dependecies are optional and may be used to enable additional features in Amino:
- Robot Models (URDF, COLLADA, Wavefront, etc.):
- GUI / Visualization:
- OpenGL
- SDL2
- plus everything under Robot Models, if you want to visualize meshes, URDF, etc.
- Raytracing (everything under Robot Models, plus):
- Motion Planning:
- FCL
- OMPL
- plus everything under Robot Models, if you want to handle meshes, URDF, etc.
- Java Bindings
- Optimization, any or all of the following:
Debian and Ubuntu GNU/Linux
Most the dependencies on Debian or Ubuntu GNU/Linux can be installed via APT. The following command should install all/most dependencies. For distribution-specific package lists, please see the files under which are used for distribution-specific integration tests.
sudo apt-get install build-essential gfortran \ autoconf automake libtool autoconf-archive autotools-dev \ maxima libblas-dev liblapack-dev \ libglew-dev libsdl2-dev \ libfcl-dev libompl-dev \ sbcl \ default-jdk \ blender flex povray ffmpeg \ coinor-libclp-dev libglpk-dev liblpsolve55-dev libnlopt-devNow proceed to Quicklisp Setup below.
Installation Errata
Some package versions are missing features or contain minor bugs that impede usage with amino. A listing of issues encountered so far is below:
OMPL Missing Eigen dependency
Version 1.4.2 of OMPL and the corresponding Debian/Ubuntu packages are missing a required dependency on libeigen. To resolve, you may need to do the following:
- Manually install libeigen: sudo apt-get install libeigen3-dev
- Manually add the eigen include directory to the ompl pkg-config file. The ompl pkg-config file will typically be in . The eigen include path will typically be . Thus, you may change the Cflags entry in to: Cflags: -I${includedir} -I/usr/include/eigen3
Blender Missing COLLADA Support
Some versions of Blender in Ubuntu and Debian do not support the COLLADA format for 3D meshes, so you may need to install Blender manually in this case (see https://www.blender.org/download/).
SBCL and CFFI incompatibility
The versions of SBCL in some distributions (e.g., SBCL 1.2.4 in Debian Jessie) do not work with new versions of CFFI. In these cases, you will need to install SBCL manually (see http://www.sbcl.org/platform-table.html).
Mac OS X
Install Dependencies via Homebrew
If you use the Homebrew package manager, you can install the dependencies as follows:
- Install packages: brew tap homebrew/science brew install autoconf-archive openblas maxima sdl2 libtool ompl brew install https://raw.github.com/dartsim/homebrew-dart/master/fcl.rb
- Set the CPPFLAGS variable so that the header can be found: CPPFLAGS=-I/usr/local/opt/openblas/include
- Proceed to Blender Setup below
Install Dependencies via MacPorts
If you use the MacPorts package manager, you can install the dependencies as follows:
- Install packages: sudo port install \ coreutils wget \ autoconf-archive maxima f2c flex sbcl \ OpenBLAS \ libsdl2 povray ffmpeg \ fcl ompl \ glpk
Ensure that or contains the MacPorts lib directory (typically ).
echo $LD_LIBRARY_PATH echo $DYLD_LIBRARY_PATHIf does not appear in either or , do the following (and consider adding it to your shell startup script).
export LD_LIBRARY_PATH="/opt/local/lib:$LD_LIBRARY_PATH"Ensure that autoconf can find the MacPorts-installed header and library files by editing the file under your preferred installation prefix (default: )
vi /usr/local/share/config.siteEnsure that the and variables in config.site contain the MacPorts directories.
CPPFLAGS="-I/opt/local/include" LDFLAGS="-L/opt/local/lib"- Proceed to Blender Setup below
Blender Setup
When installing blender on Mac OS X, you may need to create a wrapper script. If you copy the blender binaries to , then then
touch /usr/local/bin/blender chmod a+x /usr/local/bin/blender vi /usr/local/bin/blenderand add the following:
#!/bin/sh exec /usr/local/blender-2/blender.app/Contents/MacOS/blender $@
Now proceed to Quicklisp Setup below.
Quicklisp Setup
Finally, install Quicklisp manually, if desired for ray tracing and robot model compilation.
wget https://beta.quicklisp.org/quicklisp.lisp sbcl --load quicklisp.lisp \ --eval '(quicklisp-quickstart:install)' \ --eval '(ql:add-to-init-file)' \ --eval '(quit)'If you have obtained amino from the git repo, you need to initialize the git submodules and autotools build scripts.
git submodule init && git submodule update && autoreconf -iThis step is not necessary when you have obtained a distribution tarball which already contains the submodule source tree and autoconf-generated configure script.
Configure for your system. To see optional features which may be enabled or disabled, run:
./configure --helpThen run configure (adding any flags you may need for your system):
./configure- Build: make
Install:
sudo make installIf you need to later uninstall amino, use the conventional command.
The Amino distribution includes a number of demo programs. Several of these demos use URDF files which must be obtained separately.
Obtain URDF Files
- If you already have an existing ROS installation, you can install the ROS package, for example on ROS Indigo: sudo apt-get install ros-indigo-baxter-description export ROS_PACKAGE_PATH=/opt/ros/indigo/share
- An existing ROS installation is not necessary, however, and you can install only the baxter URDF and meshes: cd .. git clone https://github.com/RethinkRobotics/baxter_common export ROS_PACKAGE_PATH=`pwd`/baxter_common cd amino
Build Demos
(Re)-configure to enable demos:
./configure --enable-demos --enable-demo-baxterNote: On Mac OS X, due to dynamic loading issues, it may be necessary to first build and install amino, and the re-configure and re-build amino with the demos enabled. Under GNU/Linux, it is generally possible to enable the demos during the initial build and without installing Amino beforehand.
- Build: make
Run Demos
Many demo programs may be built. The following comand will list the built demos:
find ./demo -type f -executable \ -not -name '*.so' \ -not -path '*.libs*'The demo will lauch the Viewer GUI with a simple scene compiled from a scene file.
./demo/simple-rx/simple-scenefileSeveral demos using the Baxter model show various features. Each can be invoked without arguments.
- displays the baxter via dynamic loading
- via static linking
- moves the baxter arm in workspace
- performs collision checking
- computes an inverse kinematics solution
- computes a motion plan
- computes a motion plan to a workspace goal
- computes a sequence of motion plans
Basic Tests
- To run the unit tests: make check
- To create and check the distribution tarball: make distcheck
Docker Tests
Several Docker files are included in which enable building testing amino in a container with a clean OS installation. These Dockerfiles are also used in the Continuous Integration tests.
- To build a docker image using the file: ./script/docker-build.sh ubuntu-xenial
- To build amino and run tests using docker image built from file: ./script/docker-check.sh ubuntu-xenial
- fails with when checking for cffi-grovel.
- Older versions of SBCL (around 1.2.4) have issues with current versions of CFFI. Please try installing a recent SBCL (>1.3.4).
- I get error messages about missing .obj files or Blender being unable to convert a .dae to Wavefront OBJ.
- A: We use Blender to convert various mesh formats to Wavefront OBJ, then import the OBJ file. The Blender binaries in the Debian and Ubuntu repositories (as of Jessie and Trusty) are not built with COLLADA (.dae) support. You can download the prebuilt binaries from http://www.blender.org/ which do support COLLADA.
- When I try to compile a URDF file, I receive the error "aarx.core: not found".
- A: URDF support in amino is only built if the necessary dependencies are installed. Please ensure that you have SBCL, Quicklisp, and Sycamore installed and rebuild amino if necessary.
- When building , I get an enormous stack trace, starting with: Unable to load any of the alternatives: ("libamino_planning.so" (:DEFAULT "libamino_planning"))
This means that SBCL is unable to load the planning library or one of its dependecies, such as OMPL. Typically, this means your linker is not configured properly.
Sometimes, you just need to run or to update the linker cache.
If doesn't work, you can set the variable. First, find the location of libompl.so, e.g., by calling . Then, add the directory to your . Most commonly, this will mean adding one of the following lines to your shell startup files (e.g., .bashrc):
export LD_LIBRARY_PATH="/usr/local/lib/:$LD_LIBRARY_PATH" export LD_LIBRARY_PATH="/usr/local/lib/x86_64-linux-gnu/:$LD_LIBRARY_PATH"
- When building , I get an enormous stack trace, starting with something like: Unable to load foreign library (LIBAMINO-PLANNING). Error opening shared object "libamino-planning.so": /home/user/workspace/amino/.libs/libamino-planning.so: undefined symbol: _ZNK4ompl4base20RealVectorStateSpace10getMeasureEv.
- This error may occur when you have multiple incompatible versions of OMPL installed and the compiler finds one version while the runtime linker finds a different version. Either modify your header () and linking () paths, or remove the additional versions of OMPL.
Copyright (C) 1994, 1995, 1996, 1999, 2000, 2001, 2002, 2004, 2005, 2006, 2007, 2008, 2009 Free Software Foundation, Inc.
Copying and distribution of this file, with or without modification, are permitted in any medium without royalty provided the copyright notice and this notice are preserved. This file is offered as-is, without warranty of any kind.
Briefly, the shell commands should configure, build, and install this package. The following more-detailed instructions are generic; see the file for instructions specific to this package. Some packages provide this file but do not implement all of the features documented below. The lack of an optional feature in a given package is not necessarily a bug. More recommendations for GNU packages can be found in *note Makefile Conventions: (standards)Makefile Conventions.
The shell script attempts to guess correct values for various system-dependent variables used during compilation. It uses those values to create a in each directory of the package. It may also create one or more files containing system-dependent definitions. Finally, it creates a shell script that you can run in the future to recreate the current configuration, and a file containing compiler output (useful mainly for debugging ).
It can also use an optional file (typically called and enabled with or simply ) that saves the results of its tests to speed up reconfiguring. Caching is disabled by default to prevent problems with accidental use of stale cache files.
If you need to do unusual things to compile the package, please try to figure out how could check whether to do them, and mail diffs or instructions to the address given in the so they can be considered for the next release. If you are using the cache, and at some point contains results you don't want to keep, you may remove or edit it.
The file (or ) is used to create by a program called . You need if you want to change it or regenerate using a newer version of .
The simplest way to compile this package is:
to the directory containing the package's source code and type to configure the package for your system.
Running might take a while. While running, it prints some messages telling which features it is checking for.
- Type to compile the package.
- Optionally, type to run any self-tests that come with the package, generally using the just-built uninstalled binaries.
- Type to install the programs and any data files and documentation. When installing into a prefix owned by root, it is recommended that the package be configured and built as a regular user, and only the phase executed with root privileges.
- Optionally, type to repeat any self-tests, but this time using the binaries in their final installed location. This target does not install anything. Running this target as a regular user, particularly if the prior required root privileges, verifies that the installation completed correctly.
- You can remove the program binaries and object files from the source code directory by typing . To also remove the files that created (so you can compile the package for a different kind of computer), type . There is also a target, but that is intended mainly for the package's developers. If you use it, you may have to get all sorts of other programs in order to regenerate files that came with the distribution.
- Often, you can also type to remove the installed files again. In practice, not all packages have tested that uninstallation works correctly, even though it is required by the GNU Coding Standards.
- Some packages, particularly those that use Automake, provide , which can by used by developers to test that all other targets like and work correctly. This target is generally not run by end users.
Some systems require unusual options for compilation or linking that the script does not know about. Run for details on some of the pertinent environment variables.
You can give initial values for configuration parameters by setting variables in the command line or in the environment. Here is an example:
./configure CC=c99 CFLAGS=-g LIBS=-lposix*Note Defining Variables::, for more details.
You can compile the package for more than one kind of computer at the same time, by placing the object files for each architecture in their own directory. To do this, you can use GNU . to the directory where you want the object files and executables to go and run the script. automatically checks for the source code in the directory that is in and in . This is known as a "VPATH" build.
With a non-GNU , it is safer to compile the package for one architecture at a time in the source code directory. After you have installed the package for one architecture, use before reconfiguring for another architecture.
On MacOS X 10.5 and later systems, you can create libraries and executables that work on multiple system types–known as "fat" or "universal" binaries–by specifying multiple options to the compiler but only a single option to the preprocessor. Like this:
./configure CC="gcc -arch i386 -arch x86_64 -arch ppc -arch ppc64" \ CXX="g++ -arch i386 -arch x86_64 -arch ppc -arch ppc64" \ CPP="gcc -E" CXXCPP="g++ -E"This is not guaranteed to produce working output in all cases, you may have to build one architecture at a time and combine the results using the tool if you have problems.
By default, installs the package's commands under , include files under , etc. You can specify an installation prefix other than by giving the option , where PREFIX must be an absolute file name.
You can specify separate installation prefixes for architecture-specific files and architecture-independent files. If you pass the option to , the package uses PREFIX as the prefix for installing programs and libraries. Documentation and other data files still use the regular prefix.
In addition, if you use an unusual directory layout you can give options like to specify different values for particular kinds of files. Run for a list of the directories you can set and what kinds of files go in them. In general, the default for these options is expressed in terms of , so that specifying just will affect all of the other directory specifications that were not explicitly provided.
The most portable way to affect installation locations is to pass the correct locations to ; however, many packages provide one or both of the following shortcuts of passing variable assignments to the command line to change installation locations without having to reconfigure or recompile.
The first method involves providing an override variable for each affected directory. For example, will choose an alternate location for all directory configuration variables that were expressed in terms of . Any directories that were specified during , but not in terms of , must each be overridden at install time for the entire installation to be relocated. The approach of makefile variable overrides for each directory variable is required by the GNU Coding Standards, and ideally causes no recompilation. However, some platforms have known limitations with the semantics of shared libraries that end up requiring recompilation when using this method, particularly noticeable in packages that use GNU Libtool.
The second method involves providing the variable. For example, will prepend before all installation names. The approach of overrides is not required by the GNU Coding Standards, and does not work on platforms that have drive letters. On the other hand, it does better at avoiding recompilation issues, and works well even when some directory options were not specified in terms of at time.
If the package supports it, you can cause programs to be installed with an extra prefix or suffix on their names by giving the option or .
Some packages pay attention to options to , where FEATURE indicates an optional part of the package. They may also pay attention to options, where PACKAGE is something like or (for the X Window System). The should mention any and options that the package recognizes.
For packages that use the X Window System, can usually find the X include and library files automatically, but if it doesn't, you can use the options and to specify their locations.
Some packages offer the ability to configure how verbose the execution of will be. For these packages, running sets the default to minimal output, which can be overridden with ; while running sets the default to verbose, which can be overridden with .
On HP-UX, the default C compiler is not ANSI C compatible. If GNU CC is not installed, it is recommended to use the following options in order to use an ANSI C compiler:
./configure CC="cc -Ae -D_XOPEN_SOURCE=500"and if that doesn't work, install pre-built binaries of GCC for HP-UX.
On OSF/1 a.k.a. Tru64, some versions of the default C compiler cannot parse its header file. The option can be used as a workaround. If GNU CC is not installed, it is therefore recommended to try
./configure CC="cc"and if that doesn't work, try
./configure CC="cc -nodtk"On Solaris, don't put early in your . This directory contains several dysfunctional programs; working variants of these programs are available in . So, if you need in your , put it after.
On Haiku, software installed for all users goes in , not . It is recommended to use the following options:
./configure --prefix=/boot/commonThere may be some features cannot figure out automatically, but needs to determine by the type of machine the package will run on. Usually, assuming the package is built to be run on the same architectures, can figure that out, but if it prints a message saying it cannot guess the machine type, give it the option. TYPE can either be a short name for the system type, such as , or a canonical name which has the form:
CPU-COMPANY-SYSTEMwhere SYSTEM can have one of these forms:
OS KERNEL-OSSee the file for the possible values of each field. If isn't included in this package, then this package doesn't need to know the machine type.
If you are building compiler tools for cross-compiling, you should use the option to select the type of system they will produce code for.
If you want to use a cross compiler, that generates code for a platform different from the build platform, you should specify the "host" platform (i.e., that on which the generated programs will eventually be run) with .
If you want to set default values for scripts to share, you can create a site shell script called that gives default values for variables like , , and . looks for if it exists, then if it exists. Or, you can set the environment variable to the location of the site script. A warning: not all scripts look for a site script.
Variables not defined in a site shell script can be set in the environment passed to . However, some packages may run configure again during the build, and the customized values of these variables may be lost. In order to avoid this problem, you should set them in the command line, using . For example:
./configure CC=/usr/local2/bin/gcccauses the specified to be used as the C compiler (unless it is overridden in the site shell script).
Unfortunately, this technique does not work for due to an Autoconf bug. Until the bug is fixed you can use this workaround:
CONFIG_SHELL=/bin/bash /bin/bash ./configure CONFIG_SHELL=/bin/bashrecognizes the following options to control how it operates.
--help -h Print a summary of all of the options to `configure`, and exit. --help=short --help=recursive Print a summary of the options unique to this package's `configure`, and exit. The `short` variant lists options used only in the top level, while the `recursive` variant lists options also present in any nested packages. --version -V Print the version of Autoconf used to generate the `configure` script, and exit. --cache-file=FILE Enable the cache: use and save the results of the tests in FILE, traditionally `config.cache`. FILE defaults to `/dev/null` to disable caching. --config-cache -C Alias for `--cache-file=config.cache`. --quiet --silent -q Do not print messages saying which checks are being made. To suppress all normal output, redirect it to `/dev/null` (any error messages will still be shown). --srcdir=DIR Look for the package's source code in directory DIR. Usually `configure` can determine that directory automatically. --prefix=DIR Use DIR as the installation prefix. *note Installation Names:: for more details, including other options available for fine-tuning the installation locations. --no-create -n Run the configure checks, but stop before creating any output files. `configure` also accepts some other, not widely useful, options. Run `configure --help` for more details.Enliven: Bio analytical Techniques
Introduction
Proteins are an abundant class of large biological molecules common to all life and are the target of a huge number of biological studies. Amino acids are the major constituents of proteins and are also very common targets in biochemical studies. In certain applications, such as in the nutritional content of proteinaceous foodstuffs, it is desirable to understand the relative composition of amino acids for a certain protein or a mixture of proteins. In such applications, it is usually necessary to hydrolyze the proteins using acid hydrolysis until its individual amino acids are released and are available for detection. The most often employed acid for hydrolysis is hydrochloric acid (HCl). The advantage of HCl is that it is relatively non-oxidizing and that it can readily be removed from the hydrolysate. The classical approach of acid hydrolysis is boiling a known amount of proteins in 6 M HCl at 110°C for 18-24 h [1-3]. Following protein hydrolysis, derivatization of the amino acids is usually required for gas chromatographic analysis[4-6].
Most often, amino acid analyzers are used to profile the amino acid distribution of a protein. Commercial analyzers of this kind separate the mixture of amino acids either by ion-exchange chromatography or by high-performance liquid chromatography incorporating post-column derivatization with ninhydrin [4,5,7].but the separation times can be relatively long and detection can be hindered by complex matrix effects, especially from biological fluids like blood, urine, and spinal fluid [8,9]. Analysis using GC typically requires offline pre-column derivatization and is typically more efficient than post-column derivatization [4]. In addition, the proper choice of the derivative can improve volatility, chromatographic performance (e.g., minimizing tailing or adsorption), and improve detector sensitivity [4,10,11]. For GC, many derivatization methods have been reported with success [8,12-15]. For example, free amino acids can be derivatized directly in an aqueous solution using alkyl chloroformates, e.g., methyl, ethyl, or isobutyl. The amino acids react with propyl chloroformate and the derivates can be extracted with an organic solvent for injection directly into the GC/MS [15].
N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) is a common silylating reagent that replaces the acidic protons of the amino acids (e.g., SH, OH, NH, and COOH) with nonpolar trimethylsilane (TMS) groups. By replacing the acidic protons with nonpolar TMS groups, the polarity of an amino acid is reduced and its volatility is increased [5,13,14,16-18]. The advantage of TMS derivatization is that it requires a single step whereas other derivatization methods usually have two or more reaction steps [19,20]. The major disadvantages of TMS derivatives are their sensitivity to moisture and the derivatization of Gly and Glu that are affected by the polarity of the solvent [18,20]. For example, both the di-trimethylsilyl derivative and tri-trimethylsilyl derivatives of Gly are obtained after derivatization using either acetonitrile or acetone as solvents [18] and two peaks are observed for Glu when using chloroform as the solvent [20].
The TMS-derivatized amino acids are usually analyzed using gas chromatography/mass spectrometry (GC/MS) and identified according to a combination of their mass spectra and retention times [6,21,22]. GC/MS is a powerful instrument for the separation and identification of components in complex mixtures [23] and plays a central role in amino acid analysis by affording the advantages of improved resolution, sensitivity, and quantification, as well as faster analysis and smaller amounts of sample [1,6,16,24,25].
The previous literature contains inconsistent results about the optimal derivatization conditions of amino acids using BSTFA. These results may be related to many limitations that were observed in previous studies, such as the limited number of replicates for each of the derivatization conditions [26-29], using either peak height or peak area without evaluating the linearity of the calibration, sensitivity, or limit of detection (LOD) [18,19,27], using either TIC or SIM modes without comparing the linearity, sensitivity, or LOD, or using relatively long GC temperature programs (e.g., 54 min [27], 62 min [6], and 90 min [29]).
The use of internal standards is typically required to correct for chemical and analytical losses of amino acids during hydrolysis and derivatization. Kaspar et al. [15] reported that amino acids could be reliably quantified using 19 stable isotope-labeled amino acids as internal standards. The internal isotope standards for each amino acid would be preferable because it typically provides the best reproducibility, best accuracy and most precise result. However, internal isotope standards are very expensive. Acceptable linearity and reproducibility using norvaline (Nor) as internal standard has been reported [11] and Nor is considerably cheaper than deuterated isotope standards for each amino acid. For these reasons, Nor was selected as an internal standard for the present study.
In the present study, the method has been improved, and a comprehensive comparison of the most effective means of manipulating the chromatographic data to optimize the LODs, linearity, and sensitivity is reported. Because the mass spectrometer was operated in “fast automated scan and SIM mode” (FASST) mode, the instrument rapidly switched between TIC and SIM modes during each single injection. This capability removes the effect of any external confounding factors such as instrument drift, carryover or column bleed, which may otherwise influence the comparison of linearity and sensitivity for these two modes of operation. Moreover, this study evaluates the effects of peak area, relative peak area (i.e., referenced to the norvaline internal standard), peak height, and relative peak height (i.e., referenced to the norvaline internal standard) on the calibration figures of merit. The number of trials (9 trials) for each derivatization condition provides confidence in the statistical tests and the time needed to program the GC oven temperature (16 min) is considerably shorter than those reported by others. In addition, the method was reproducible even when data was collected across a span of 60 days.
During the acid hydrolysis step, the majority of the amino acids are unaltered. However, tryptophan (Trp) and cystine (Cyt) are decomposed. Methionine (Met) undergoes oxidation. Glutamine (Gln) and asparagine (Asn) are typically deamidated to aspartic acid (Asp) and glutamic acid (Glu), respectively [1]. In addition, arginine (Arg) decomposes to ornithine during silylation, which can yield inaccurate values in the analysis of free amino acids in extracts of biological fluids as well as cell and tissue extracts [1]. Loosing histidine (His) and Arg amino acids after acid hydrolysis was also reported by Rayner [30]. Therefore, the optimization process evaluated linearity, accuracy, LOD, and limit of quantitation (LOQ) for the 14 remaining amino acids. These amino acids were also selected because they comprise the majority of the amino acids in human hair, which is the intended application for this method. Additionally, these amino acids will be easiest to measure by GC/MS, because they will offer the best signal to noise (S/N) values, and are least affected by co-elution with smaller constituents [29]. Because there is no known hair standard available with which to validate our method, horse heart myoglobin was used to validate the method. Although myoglobin does not contain disulfide bonds, it does contain a high degree of alpha-helical structure, much like the keratins found in hair. The method has been used on dozens of hair samples and has provided adequate within-person variance results when separate aliquots of hair from the same individual are analyzed. Results of the hair analyses are reported in two separate manuscripts, one will submit to the forensic international journal and the second will submit to the diabetes journal.
Materials and Methods
Reagents and Supplies
The 14 amino acids of interest in this study are L-alanine (Ala), L-glycine (Gly), L-Valine (Val), L-leucine (Leu), L-isoleucine (Ile), L-proline (Pro), L-serine (Ser), L-threonine (Thr), L-aspartic acid (Asp), L-glutamic acid (Glu), L-phenylalanine (Phe), L-lysine (Lys), L-tyrosine (Tyr), and L-cystine (Cyt). L-norvaline (Nor) was used as an internal standard. All these amino acid standards were purchased from Sigma-Aldrich (St. Louis, MO, USA). The derivatizing agent was N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) (Supelco Analytical, Bellefonte, PA, USA). Hydrochloric acid (HCl) 6 M was used as the hydrolysis agent to liberate free amino acids from horse heart myoglobin. Acetonitrile, methanol, acetone, and chloroform were purchased from GPS Chemicals (Columbus, OH, USA). Horse heart myoglobin was also purchased from Sigma-Aldrich (St. Louis, MO, USA).
This study used 4-mL glass vials with phenolic rubber lined caps (Qorpak, Bridgeville, PA, USA). Solutions were filtered through a 13 mm × 0.45 μm, polyvinylidene difluoride (PVDF) filter (Bonna-Agela Technologies, Wilmington, DE, USA). The nitrogen generator was purchased from Parker Hannifin Corporation (Haverhill, MA, USA).
Standard Stock Solutions
Standard amino acid stock solutions were prepared by dissolving each of the 14 amino acids in 0.1 M HCl to a concentration of 5 μmol/mL. An internal standard stock solution was prepared by dissolving Nor in 0.1 M HCl to a concentration of 10 μmol/mL. The solutions were stored at 4°C until analysis. Calibration standards at 7 different concentrations (0.001-0.3 μmol/mL were prepared using the 14 standard amino acid stock solutions. In each solution, the concentration of internal standard Nor was 0.16 μmol/mL.
Amino Acid Hydrolysis
Determining the amino acid content of myoglobin involved the acid hydrolysis in 6 M HCl for 24 h until the protein-bound amino acids were released and were available for detection. The myoglobin sample was separated into three subsamples and precisely weighed to approximately 3.0 mg and transferred into a 4-mL glass vial with a phenolic rubber lined cap. Thereafter, 1.0 mg of Nor was added. Each subsample was hydrolyzed by adding 0.3 mL of 6M HCl to the vial and was tightly capped. To heat the samples, an aluminum block with holes to accommodate the glass vials was mounted on a hot-plate for 24 h at 110°C. After cooling the solution to room temperature, the solutions were filtered and dried under a constant 4 L/min nitrogen stream.
Amino Acid Derivatization
4.4.1) Effect of Solvent: A 100 μL aliquot of the 0.33 μmol/mL standard amino acid solution was pipetted into a 4-mL glass vial and dried under a nitrogen stream. The existence of moisture can result in poor reaction yield and instability of the derivatized products. The dried amino acid residues were dissolved in a 100-μL aliquot of acetonitrile and derivatized with the addition of a 100-μL aliquot of BSTFA. The glass vial was tightly capped, ultrasonicated for 1 min, and mounted on a hot-plate for 30 min at 100°C. For the solvent study, the only change made was to substitute the acetonitrile with chloroform and acetone. Triplicate samples for each solvent were prepared and replicated three times to yield 9 measurements for each solvent.
Effect of Time and Temperature Reactions
Standard solutions of 0.33 μmol/mL were prepared and stored at 4 °C. A 100-μL aliquot of this solution was transferred into a 4-mL glass vial and dried under constant nitrogen stream. The dried amino acid residues were dissolved in a 100-μL aliquot of acetonitrile and derivatized with the addition of a 100-μL aliquot of BSTFA. The glass vial was tightly capped, ultrasonicated for 1 min and mounted on a hot-plate at 50, 100, or 150°C with reaction times of 15, 30, 45, or 60 min, separately. Precision was measured by using Triplicate samples for each time at each temperature were prepared and replicated three times to yield 9 measurements for each time at each temperature.
GC/MS Analysis
Amino acid derivatives were analyzed with a Shimadzu GC/MS instrument (QP-2010SE, Scientific Instrument, Inc. Columbia, MA, USA). Amino acids were separated on 5% diphenyl-dimethylpolysiloxane capillary column (SHRX1-5MS, 30 m X 0.25 mm X 0.25 μm). Ultra-high purity helium was used as a carrier gas at a constant flow rate of 21.5 mL/min. The transfer column flow was 1.0 mL/min.
The temperature of the column was programmed to rise from 70°C to 170°C at a rate of 10°C/min, and then was ramped to 280°C at a rate of 30°C/min, at which it was held for 3 min. The total run time was 16.6 min. Sample injection was performed in split injection mode (1:20 ratio) at 280°C using an injection volume of 1 μL. Triplicates injections for each sample were run in a random block design (i.e., complete set of samples analyzed in randomized order, before the next set of sample replicates are analyzed in a rerandomized order) using acetonitrile as a solvent blank before each injection. Three solvent vials of acetonitrile, methanol, and acetone were used sequentially as cleaning solvents for the autosampler injection syringe.
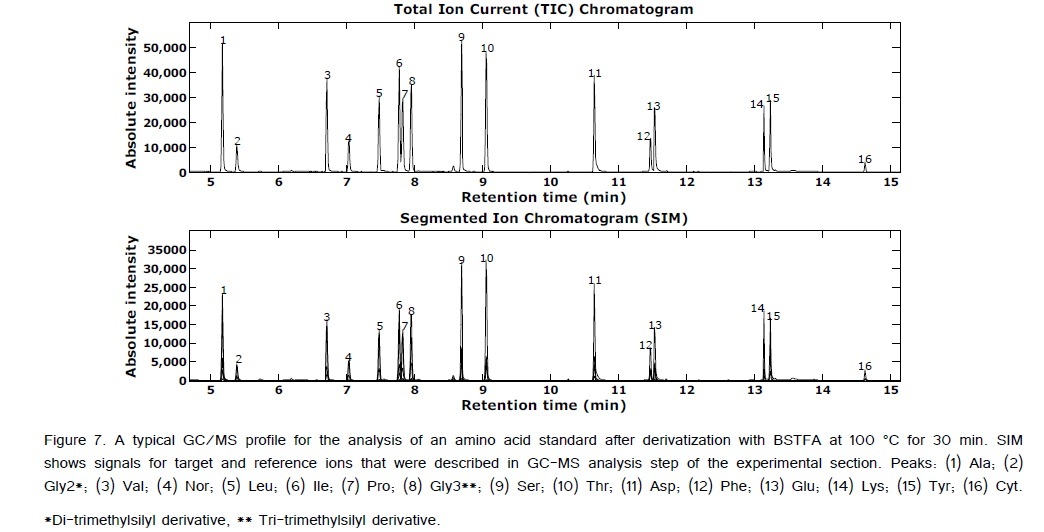
The mass spectrometer was operated in fast automated scan and SIM type (FASST) mode, which switches between full scan mode and selected ion monitoring during a single analysis. The mass spectrometer was operated in full scan mode (TIC) from m/z of 50 to m/z of 500 with a scan time of 0.3 s, and selected ion monitoring mode (SIM) with a scan time of 0.2 s. For SIM, the appropriate ion set of one target ion and two reference ions as characteristics mass fragments of the derivatized amino acid was used. Selection of the characteristic ions for the quantification ion is very important. The target ions are the fragment ions of the highest intensity (base peak), while the reference ions are two other fragment ions of the next greatest intensities [27].Table 1 presents the target and reference ions that were quantify as a set of each amino acid.
| Amino acid | Target ion (m/z) | Reference ions (m/z) |
Ala | 116 | 73, 147 |
Gly of di-trimethylsilyl derivative | 102 | 73, 147 |
Val | 144 | 73, 218 |
Nor | 144 | 73, 218 |
Leu | 158 | 73, 102 |
Ile | 158 | 73, 218 |
Pro | 142 | 73, 216 |
Gly of tri-trimethylsilyl derivative | 174 | 73, 248 |
Ser | 304 | 218, 73 |
Thr | 73 | 218, 291 |
Asp | 232 | 156, 73 |
Glu | 246 | 73, 128 |
Phe | 218 | 192, 73 |
Lys | 174 | 156, 73 |
Tyr | 218
| 179, 280
|
Cyt | 218 | 147, 73 |
Table 1. Target and reference ions for SIM mode as characteristics mass fragments of the 14 amino acids.
Acid Hydrolysis Effects on Free Amino Acids
Triplicate standard amino acid solutions were prepared by dissolving approximately 0.1 mg of each amino acid and Nor in 1 mL of 0.1 M HCl. The aqueous mixture was split into two glass vials and dried under a constant nitrogen stream. One half was derivatized and analyzed by GC/MS, whereas the other half was hydrolyzed using 1 mL of 6 M HCl, derivatized, and analyzed.
Calibration Curves and Reproducibility
To evaluate the linearity and sensitivity of the signal with respect to concentration, eight linear calibrations were generated for each amino acid. The first set of calibrations modeled the absolute peak areas of the corresponding amino acids with respect to their amino acid concentrations (μmol/mL). The second set of calibrations modeled the relative peak areas referenced to the Nor internal standard. The third set of calibrations modeled the absolute peak heights with respect to the amino acid concentrations. The fourth set of calibrations modeled the relative peak heights referenced to the Nor internal standard. These four calibrations were obtained from the total ion chromatograms and the extracted ion chromatograms (using the target/quant ions reported in the section above) to yield a total of eight calibrations.
Data Analysis
Statistical analyses were performed using MATLAB R2012b or R2014a (MathWorks, Natick, MA). Excel 2010 (Microsoft, Redmond, WA) was also used to calculate the concentrations and calibrations. All statistical tests were conducted at the 95% confidence level.
Results and Discussion
Method validation included the evaluation of the independent procedures of acid hydrolysis, amino acid derivatization, and GC/MS analysis. To fully assess the performance of these procedures, a standard amino acid mixture enabled the verification of the GC/MS method, including derivatization and acid hydrolysis effects on amino acids, while the reference protein sample (i.e., horse heart myoglobin) helped assess the effectiveness of acid hydrolysis. In addition, any variation in conditions from one run to the next was controlled by referencing all data to the Nor internal standard peak [5,11,15,31].
Effect of Solvent on Derivatization
Analysis of variance-principal component analysis (ANOVA-PCA) evaluated the sources of variation arising from the differences among the relative peak areas (i.e., referenced to the Nor internal standard) of the amino acids and the three different solvents that were used for derivatization [32]. As presented in Figure 1, the 3 solvents formed well resolved clusters of principal component scores. The ellipses around the scores are 95% CI and the effects of all three solvents were significant. The first two principal components account for 89% of the total variance. The first principal component separated the acetonitrile from the other two solvents. Hence, the variable loadings of this principal component will indicate the overall effect of the solvent. Figure 2 are the variable loadings of the first component; positive loading correspond to amino acid peaks that are larger for the acetonitrile solvent and negative loadings correspond to amino acid peaks that are larger for one of the other two solvents.
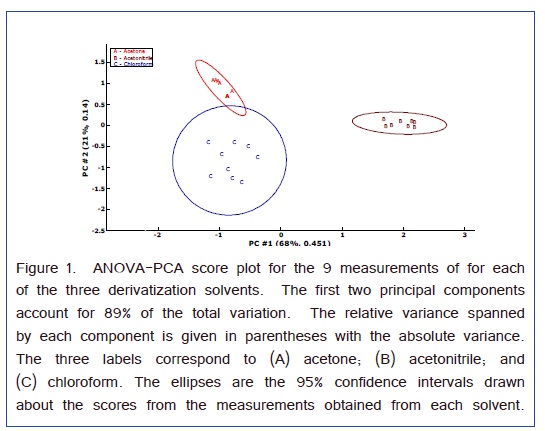
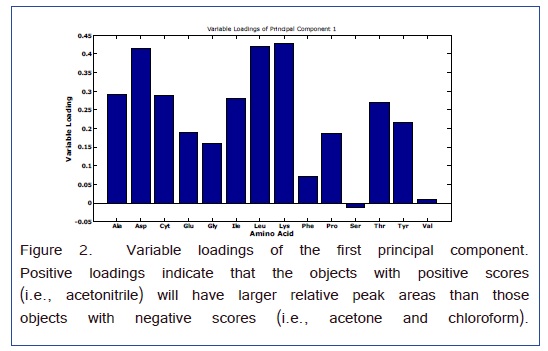
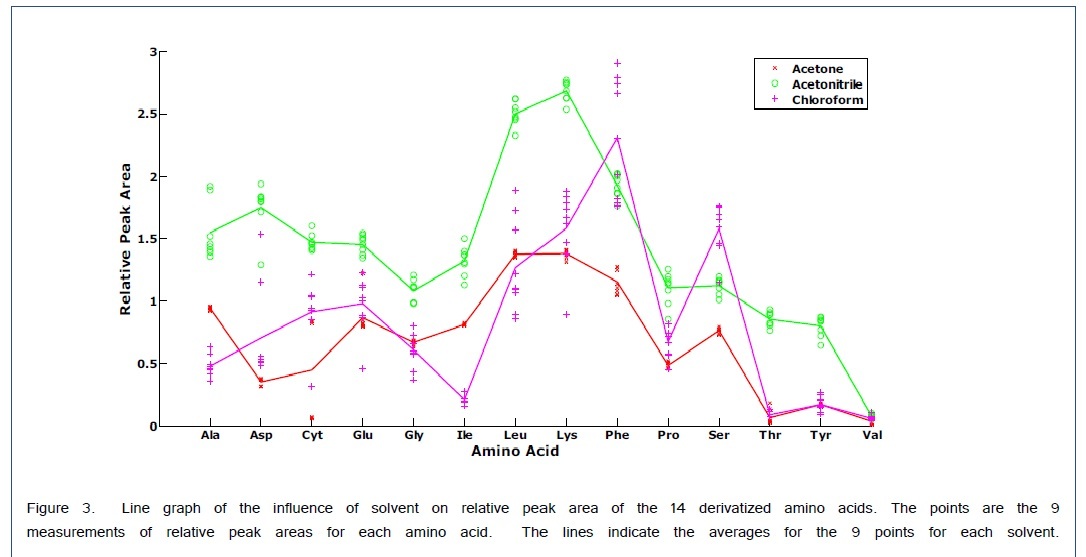
A scatter plot with the superimposed averages gives the relative peak areas for each amino acid with respect to the solvent in Figure 3. This graph is consistent with the variable loadings plot in that chloroform gives a larger Ser peak area than that obtained from acetonitrile. For the Phe peak, although the average is larger for chloroform the relative peak areas are much less precise which gave a lower weight for the variable loading in Figure 2. Therefore, the Phe variable loading on the first PC is positive indicating a favorable result for acetonitrile as a solvent. For all the other amino acids, acetonitrile appears to work the best.
The derivatization of Gly and Glu were affected by the polarity of the solvent. Sharp single peaks were obtained for all amino acids except Gly, in which case both the di-trimethylsilyl derivative and tri-trimethylsilyl derivative were obtained after derivatization using acetonitrile or acetone as solvents.In contrast to these solvents, two peaks were observed for Glu when using chloroform as the solvent. Ala, Val, Nor, Leu, Ile, Pro, and Phe produced di-trimethylsilyl derivatives in all solvents and Ser, Thr, Asp, Glu, Lys, and Tyr yielded tri-trimethylsilyl derivatives in all solvents. Cyt formed a tetra-trimethylsilyl derivative. These results are similar to those obtained by Cehrke et al. [20] who also found that the derivatization of Gly and Glu were affected by the polarity of the solvent. For less polar solvents such as chloroform, only one peak was obtained for glycine and two peaks were obtained for Glu. Conversely with more polar solvents such as acetonitrile and acetone, one observes two peaks for Gly and only one peak for Glu. The observation of two products for certain amino acids, although not ideal, does not necessarily prevent quantitation. If both products are chromatographically resolved, the sum of the two peak heights or peak areas can be used for quantitation.
Effect of Temperature and Time on Derivatization
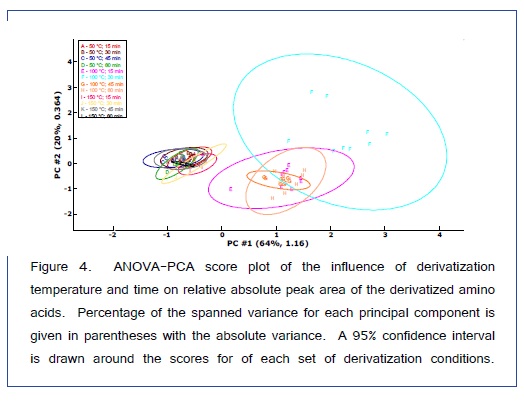
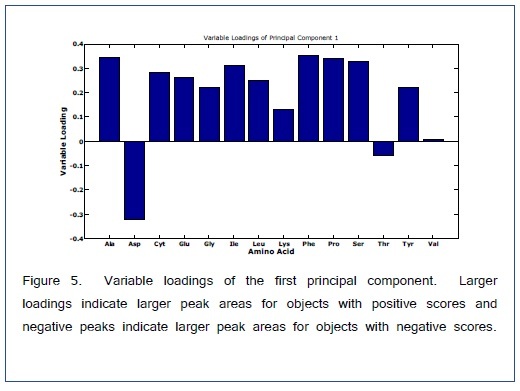
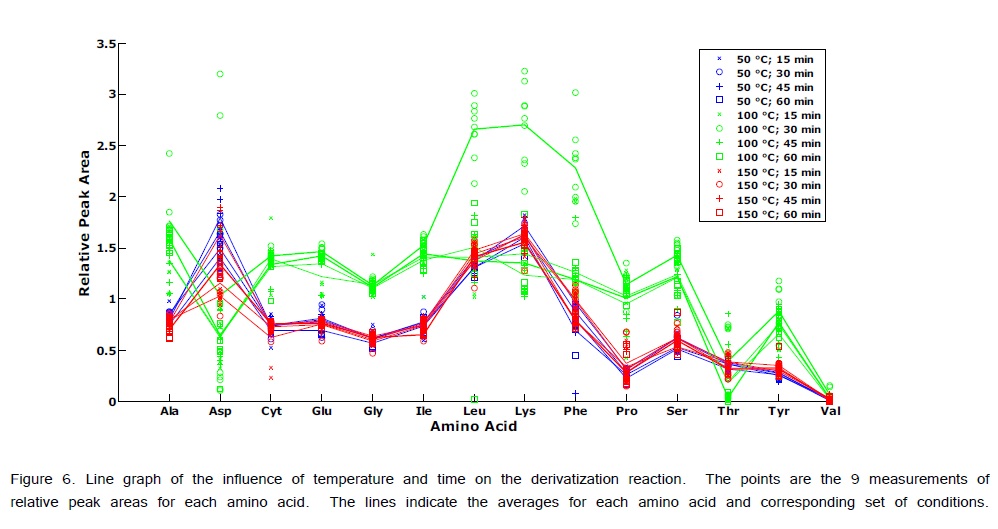
ANOVA-PCA explored the effects of different temperatures (50, 100, and 150°C) and different times (15, 30, 45, and 60 min) on the relative peak areas for the 14 amino acid derivatives. Figure 4 is the PCA score plot, all scores for the reaction temperature of 100°C are positive on the first principal component. The most extreme cluster of scores is for 100°C and 30 min. The first two principal components account for 84% of the total variance. The variable loadings of the first component (Figure 5) and the scatter plot in Figure 6, the trend of all amino acids are positively enhanced using the reaction temperature of 100°C for 30 min, except for Asp and Thr amino acids Consequently, the reaction temperature of 100 °C for 30 min has the largest product yield. This result is in agreement with Shen et al. [24] and Deng et al. [33] who concluded that the optimal derivatization conditions were acetonitrile as a solvent, a temperature of 100 °C, and a reaction time of 30 min. Figure 7 is a typical GC/MS profile for the standard amino acid mixture at a concentration of 0.3 μmol/mL that was obtained under these optimal conditions.
Total ion Current Mode (TIC) and Selected Ion Monitoring Mode (SIM)
Paired sample t-tests were conducted to compare SIM and TIC modes using different parameters: the coefficient of determination (R2) and the limit of detection (LOD) of the calibration line. The concentration LOD is the lowest concentration of an analyte in a sample that can be detected above the backgrounds, but not necessarily quantified [34]. The LOD was calculated by dividing three times the standard error of the calibration line at the intercept by the slope of the calibration line [34]. First, the absolute peak areas and the relative peak areas (i.e., referenced to the Nor internal standard) for each mode (SIM or TIC) were compared separately. The R2 and LOD values were significantly different between the absolute and relative peak area calibrations for both SIM and TIC acquisition modes. The absolute and relative peak heights (i.e., referenced to the Nor internal standard) for each analysis mode were also compared.
The R2 and LOD values were significantly different between the absolute and relative peak height calibrations. Based on these results, the relative peak area and the relative peak heights are preferred to the absolute values for both SIM and TIC modes of data acquisition, as presented in Table 2. R2 and LOD are the average of R2 and LODs of the 14 amino acids. Moreover, the minimum and maximum values of LOD indicate that the relative values are always more sensitive than the absolute values. Second, the relative peak area (i.e., referenced to the Nor internal standard) and the relative peak heights (i.e., referenced to the Nor internal standard) were compared between the two acquisition modes (Table 3). The significant difference noted in relative peak area. The R2 and LOD in SIM mode of relative peak area is better than TIC mode, as well as the lower minimum and maximum values.
| Calibration parameters | SIM (n=14) | TIC (n=14) | ||||||||||
| Absolute Area | Relative Area | P-value | Absolute Height | Relative Height | P-value | Absolute Area | Relative Area | P-value | Absolute Height | Relative Height | P-value | |
| Mean ± 95%CI | Mean ± 95%CI | Mean ± 95%CI | Mean ± 95%CI | Mean ± 95%CI | Mean ± 95%CI | Mean ± 95%CI | Mean ± 95%CI | |||||
R2 | 0.95 | 0.993 | 0.001 | 0.94 | 0.97 | 0.002 | 0.95 | 0.97 | 0.01 | 0.95 | 0.97 | 0.02 |
± 0.02 | ± 0.003 | ± 0.03 | ± 0.01 | ± 0.03 | ± 0.02 | ± 0.01 | ± 0.02 | |||||
LOD (µmol/L) | 0.09 ± 0.04 | 0.06 ± 0.02 | 0.04 | 0.13 ± 0.09 | 0.07 ± 0.01 | 0.03 | 0.14 ± 0.08 | 0.07 ± 0.01 | 0.01 | 0.09 ± 0.04 | 0.07 ± 0.02 | 0.04 |
Min LOD | 0.05 | 0.04 | 0.05 | 0.05 | 0.06 | 0.07 | 0.08 | 0.07 | ||||
Max LOD | 0.3 | 0.1 | 0.9 | 0.1 | 0.9 | 0.1 | 0.9 | 0.1 | ||||
Table 2. The comparison of absolute peak area, relative peak area, absolute peak height, and relative peak height in SIM and TIC modes, the mean is the average of the 14 amino acids. A matched-sample t-test was used to determine the p-values.
| Calibration parameters | SIM | TIC | p-values |
| Mean ± 95% CI (n=14) | Mean ± 95% CI (n=14) | ||
R² of relative peak area | 0.993 ± 0.003 | 0.97 ± 0.02 | 0.04 |
LOD of relative peak area | 0.06 ± 0.02 | 0.07 ± 0.01 | 0.05 |
Min LOD | 0.04 | 0.07 | |
Max LOD | 0.1 | 0.1 | |
R² of relative peak height | 0.97 ± 0.01 | 0.97 ± 0.02 | 0.93 |
LOD of relative peak height | 0.07 ± 0.03 | 0.07 ± 0.01 | 0.4 |
Min | 0.05 | 0.07 | |
Max | 0.1 | 0.1 |
Table 3. Comparison of average R2 values and LOD values of relative peak area and relative peak height of the 14 amino acids in SIM and TIC modes. A matched sample t-test was used to determine the p-values.
Third, paired sample t-tests were also conducted to compare R2 of relative peak areas and relative peak heights within the SIM mode. The R2 was significantly larger for relative peak area than for relative peak height (p= 0.02). According to these results, the coefficient of determination of the relative peak area is better than that of the relative peak height.
To summarize, the internal standard (Nor) improved the coefficient of determination for both MS modes, and peak area provided better performance than peak height for the TIC and SIM modes. The SIM mode had better sensitivity and linearity than the TIC mode. The best method was therefore SIM mode, with the peak areas normalized to the internal standard, Nor. The calibration curve results for the relative peak areas for the 14 amino acids studied are reported in Table 4.
| Retention time (min) | Regression line (n=3) | Limit of detection (µmol/L) | Limit of | |||
| Amino acid | quantitation | |||||
| R2 ± 95% CI | Slope ± 95% CI | Intercept ± 95% CI | (µmol/L) | |||
Ala | 5.19 | 0.991 ± 0.003 | 5.9 ± 0.2 | 0.042 ± 0.007 | 0.07 | 0.3 |
Gly | 5.36 | 0.99 ± 0.01 | 8.7 ± 0.7 | 0.04 ± 0.04 | 0.04 | 0.2 |
Val | 6.73 | 0.991 ± 0.003 | 6.9 ± 0.2 | 0.031 ± 0.005 | 0.04 | 0.2 |
Leu | 7.85 | 0.994 ± 0.003 | 5.3 ± 0.1 | 0.03 ± 0.02 | 0.07 | 0.2 |
Ile | 7.79 | 0.993 ± 0.002 | 6.5 ± 0.2 | 0.031 ± 0.005 | 0.04 | 0.1 |
Pro | 7.84 | 0.995 ± 0.002 | 8.5 ± 0.1 | 0.021 ± 0.009 | 0.04 | 0.1 |
Ser | 8.71 | 0.996 ± 0.009 | 9.8 ± 0.2 | 0.008 ± 0.007 | 0.04 | 0.1 |
Thr | 9.07 | 0.993 ± 0.006 | 12.7 ± 0.7 | 0.04 ± 0.09 | 0.08 | 0.2 |
Asp | 10.66 | 0.997 ± 0.002 | 15.9 ± 0.9 | -0.09 ± 0.07 | 0.05 | 0.1 |
Glu | 11.48 | 0.99 ± 0.01 | 5.03 ± 1.27 | -0.176 ± 0.007 | 0.04 | 0.3 |
Phe | 11.54 | 0.996 ± 0.009 | 9.6 ± 0.7 | -0.07 ± 0.06 | 0.07 | 0.2 |
Lys | 13.15 | 0.99 ± 0.02 | 4.5 ± 0.3 | -0.11± 0.01 | 0.08 | 0.2 |
Tyr | 13.24 | 0.995 ± 0.003 | 9.5 ± 0.5 | -0.24 ± 0.09 | 0.08 | 0.2 |
Cyt | 14.64 | 0.97 ± 0.01 | 1.3 ± 0.4 | -0.2 ± 0.1 | 0.1 | 0.5 |
Table 4. Retention time, R2 values, calibration equation, limit of detection (LOD), and limit of quantitation (LOQ) of the derivatized amino acids using SIM mode after normalization to the internal standard, Nor.
Limits of Detection and Quantitation
The limit of quantitation (LOQ) is defined as the concentration that gives a signal to noise (S/N) ratio of 10 [11,15] whereas the LOD is usually defined as the concentration at which the signal to noise ratio is 3 [11,15,24]. In Table 4, the LODs were calculated by dividing three times the standard error of the calibration line at the intercept by the slope of the calibration line and the LOQs were calculated by dividing 10 times the standard error of the calibration line at the intercept by the slope of the calibration line. As tabulated in Table 4, for liquid standard injections, the LODs and the LOQs were in the ranges of 0.04-0.1 μmol/L and 0.1-0.5 μmol/L, respectively, depending on the amino acid under consideration.
Bartolomeo et al. [11] reported less sensitive LOQs than those reported in current method (3-16 μmol/L) for BSTFA-derivatized amino acids. Kasper et al. [15] also reported less sensitive LOQs of 0.3-30 μmol/L and LODs (0.03-0.12 μmol/L) for propyl chloroformate-derivatized free amino acids in biological fluids. Whereas, Shen et al.[24] reported similar LODs of 0.08-0.7 μmol/L for BSTFA-derivatized amino acids.
Acid Hydrolysis Effects on Free Amino Acids
Acid hydrolysis is a crucial step that influences amino-acid recovery. During acid hydrolysis, most (nine) of the amino acids were unaffected. However, Gln and Asn are typically deamidated to Asp and Glu, respectively. Arg decomposes to ornithine during silylation, which can yield inaccurate values in the analysis of free amino acids in extracts of biological fluids as well as cell and tissue extracts. Cyt totally decomposed. Ser, Tyr, and Thr were also partially lost due to their lability during acid hydrolysis [2,3,25].
The calculated recoveries were based on the measured weight of the amino acids in the starting solutions and the calculated mass based from the relative peak areas. To calculate the original amino acid mass from the peak area, external calibrations were used liquid standards, the known GC split, the known injection volume, and solvent dilution factors. The liquid calibration modeled the relative peak area of each amino acid with respect to the corresponding prepared concentrations. Amino acid standard in Table 5 reports the amino acids that have been recovered after acid hydrolysis.
| Amino acid | Actual weight (mg) | Calculated weight via deriv. GC/MS (mg) | Recovery (%) | Calculated weight after acid hydrolysis and deriv. GC/MS (mg) | Recovery (%) |
| Mean ± 95% CI | Mean ± 95% CI | Mean ± 95% CI | Mean ± 95% CI | Mean ± 95% CI | |
| (n=3) | (n=9) | (n=9) | (n=9) | (n=9) | |
Ala* | 0.12 ± 0.01 | 0.14 ± 0.02 | 118 ± 2 | 0.13± 0.03 | 105* ± 2 |
Gly | 0.13 ± 0.02 | 0.12 ± 0.02 | 97 ± 2 | 0.07 ± 0.01 | 72 ± 1 |
Val* | 0.078 ±0.004 | 0.08 ± 0.01 | 110 ± 1 | 0.08 ± 0.01 | 109* ± 7 |
Leu* | 0.147 ±0.002 | 0.14 ± 0.01 | 93 ± 6 | 0.16 ± 0.02 | 105* ± 3 |
Ile* | 0.19 ± 0.02 | 0.17 ± 0.03 | 89 ± 3 | 0.18 ± 0.04 | 88* ± 3 |
Pro* | 0.16 ± 0.02 | 0.14 ± 0.02 | 99 ± 4 | 0.15 ± 0.02 | 108* ± 1 |
Ser | 0.16 ± 0.03 | 0.18 ± 0.04 | 103 ± 4 | 0.12 ± 0.04 | 70 ± 4 |
Thr | 0.21 ± 0.04 | 0.21 ± 0.03 | 98 ± 2 | 0.15 ± 0.03 | 75 ± 2 |
Asp* | 0.16 ± 0.03 | 0.16 ± 0.04 | 105 ± 3 | 0.19 ± 0.02 | 103* ± 1 |
Glu* | 0. 18 ± 0.02 | 0.21± 0.04 | 106 ± 2 | 0.16 ± 0.03 | 92* ± 1 |
Phe* | 0.22 ± 0.01 | 0.22 ± 0.03 | 103 ± 1 | 0.19 ± 0.04 | 91* ± 2 |
Lys* | 0.18 ± 0.02 | 0.21 ± 0.02 | 111 ± 1 | 0.13 ± 0.01 | 93* ± 5 |
Tyr | 0.21 ± 0.02 | 0.23 ±0.02 | 110 ± 1 | 0.14 ± 0.1 | 73 ± 4 |
Cyt | 0.29 ± 0.03 | 0.3 ± 0.1 | 102 ± 3 | ||
Average recovery (%) | 103 | 99* |
* Average of the best-recovered amino acids: Ala, Leu, Phe, Asp, Glu, Val, Ile, Pro, and Lys.
Table 5. Amino acid recovery (%) calculated from single amino acids.
Recoveries after the acid hydrolysis step were 70, 74, and 75% for Ser, Tyr, and Thr, respectively. As discussed above, Gly is known to give two derivatives after the BSTFA reaction in polar solvents, so was not included in the acid-hydrolysis validation study. Therefore, the validation parameters were estimated using the nine best-recovered amino acids: Ala, Leu, Phe, Asp, Glu, Val, Ile, Pro, and Lys. These 9 amino acids all were recovered within the accepted range 80-110% [35]. Recoveries of the best amino acids after acid hydrolysis ranged from 88.9% for Ile up to 109.8% for Val, while the average recovery was 99.7%, which is within 80-110% that is widely considered acceptable [35].
Accuracy
A myoglobin standard with a defined distribution of amino acids was hydrolyzed and subjected to the analysis. This evaluation was important because amino acids differ in the ease with which they are liberated from peptide linkages during acid hydrolysis. Amino acids such as Val and Ile are linked by peptides bonds that are not easily broken [2,3,25,36]. Val and Ile values were lower than expected because of incomplete hydrolysis; peptides with an Ile-Val pair in their sequence are known to be resistant to acid hydrolysis because of steric effects [2].
Table 6 reports the results of the myoglobin experiment. As expected, Cyt appeared to be completely hydrolyzed following acid hydrolysis [2,3,11,25]. Ser, Tyr, Pro, and Thr are completely liberated after hydrolysis for 20 h, but they progressively decompose under acid hydrolysis [2,3,11,25,31]. Moreover, because Gln and Asn were converted to Glu and Asp [31], Table 6 reports the actual and measured Asp mole fractions. The measured mole fractions for myoglobin Asp was obtained from sum of Asp and Asn. The sum of the mole fractions for Glu and Gln are also reported. Additionally, based on the acid hydrolysis effects on free amino acids presented in Table 5, the validation parameters were estimated using the best-recovered amino acids: Ala, Val, Leu, Ile, Phe, Asp, Glu, Pro, and Lys. The TIC and SIM Chromatograms of the amino acids obtained from myoglobin are given in Figure 8.
Based on these results, the relative peak areas of the SIM mode gave the better performance. The measured mole fractions of myoglobin amino acids were obtained by: (1) the concentration (μmol/mL) for each amino acid was calculated using external calibration curves of liquid standards, (2) the amino acid concentrations from the calibration curves were used to calculate the moles for each amino acid, (3) the total moles of analyzed amino acids were calculated, (4) the molar ratios were calculated by dividing the measured moles for each amino acid by the total experimental moles, and (5) multiplying fraction of the amino acid by 100 expresses the mole fraction as a percentage.
| Amino acid | True mole percent obtained from sequence | Experimently obtained mole percent | Absolute error | True mole percent of the nine best recovered amino acids obtained from sequence | Experimentaly obtained | Absolute error (mole percent) |
| Mean ± 95% CI | mole percent of the nine best recovered amino acids# | |||||
| n=9 | Mean ± 95% CI | |||||
| n=9 | ||||||
Ala | 11.2 | 13 ± 1 | 2.2 | 14.2 | 15 ± 1 | 0.8 |
Gly | 11.2 | 11 ± 1 | -0.2 | |||
Val | 5.1 | 3.4 ± 0.3 | -1.7 | 6.5 | 5.1 ± 0.3 | -1.4 |
Leu | 12.5 | 15 ± 1 | 2.5 | 15.8 | 18 ± 1 | 2.2 |
Ile | 6.6 | 6 ± 1 | -0.6 | 8.4 | 7 ± 1 | -1.4 |
Pro | 2.9 | 4 ± 1 | 1.1 | 3.7 | 4.3 ± 0.2 | 0.6 |
Ser | 3.7 | 1.1 ± 0.1 | -2.6 | |||
Thr | 5.1 | 2.4 ± 0.1 | -2.7 | |||
Asp* | 7.4 | 9 ± 1 | 1.6 | 9.3 | 11 ± 1 | 1.7 |
Glu** | 13.9 | 15 ± 1 | 1.1 | 17.8 | 18 ± 1 | 0.2 |
Phe | 5.1 | 5.4 ± 0.4 | 0.3 | 6.5 | 6.4 ± 0.4 | -0.1 |
Lys | 13.9 | 13.1 ± 0.4 | -0.8 | 17.8 | 18 ± 1 | 0.2 |
Tyr | 1.4 | 0.4 ± 0.2 | -1 | |||
Total | 100 | 100 | -0.8 | 100 | 100 | 2.8 |
* Asp is the sum of Asn and Asp.
** Glu is the sum of Gln and Glu.
# The best-recovered amino acids: Ala, Leu, Phe, Asp, Glu, Val, Ile, Pro, and Lys.
Table 6. Measured mole percentage of amino acid composition for myoglobin protein and true mole percentage of amino acids obtained from the sequence.
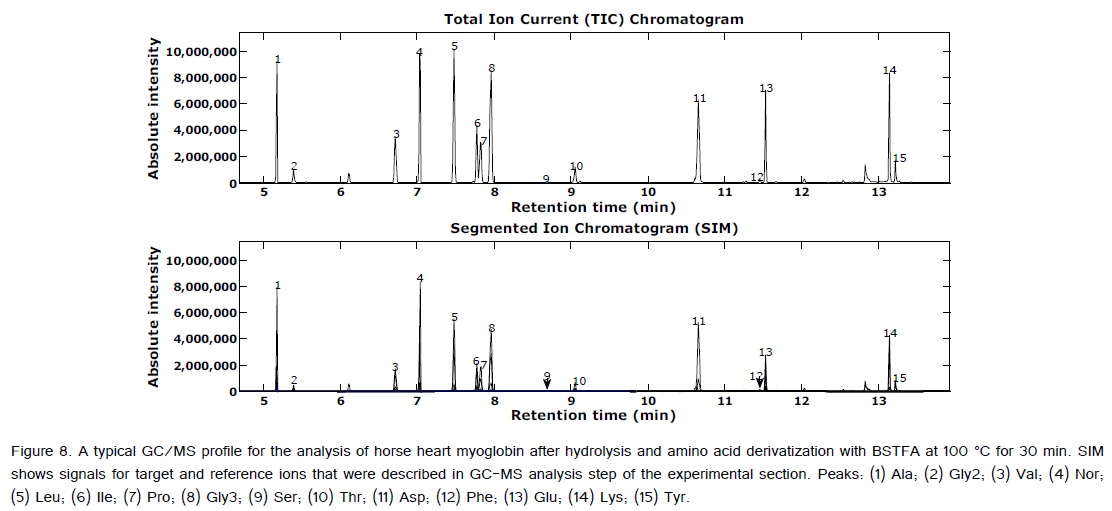
Conclusion
The presented method was successfully developed for the determination of amino acids in protein. BSTFA derivatization with GC/MS analysis offered a balanced compromise of features over other approaches of amino acid analysis. The derivatization is a single step procedure, the total GC/MS analysis time is 16.6 min, and the MS detection software enabled FASST mode switching between full scan mode and selected ion monitoring mode during a single analysis. Real-time switched-mode data acquisition enabled a direct comparison between the different modes of operation (SIM versus TIC). Peak areas provided lower LODs than peak heights for TIC and SIM modes. The SIM mode had better LODs than the TIC mode, as expected. The best method was therefore SIM mode with the peak area normalized to the norvaline internal standard. The analytical procedures demonstrated in this paper are expected to be applicable to other proteins, and have been reproducible with hair analysis [29].
In addition, ANOVA-PCA was applied for the first time for optimizing the derivatization of a complex set of samples. This method can simplify the optimization when there are many response variables.
Acknowledgements
What’s New in the Amino App Pc Download Archives?
Screen Shot

System Requirements for Amino App Pc Download Archives
- First, download the Amino App Pc Download Archives
-
You can download its setup from given links:

Amino App Pc Download Archives & PC Free Download

Amino App Pc Download Archives& Crack
